

Biallelic Variants in the Ectonucleotidase ENTPD1 Cause a Complex Neurodevelopmental Disorder with Intellectual Disability, Distinct White Matter Abnormalities, and Spastic Paraplegia
- PMID: 35471564
- PMCID: PMC10054521
- DOI: 10.1002/ana.26381
Biallelic Variants in the Ectonucleotidase ENTPD1 Cause a Complex Neurodevelopmental Disorder with Intellectual Disability, Distinct White Matter Abnormalities, and Spastic Paraplegia
Abstract
Objective: Human genomics established that pathogenic variation in diverse genes can underlie a single disorder. For example, hereditary spastic paraplegia is associated with >80 genes, with frequently only few affected individuals described for each gene. Herein, we characterize a large cohort of individuals with biallelic variation in ENTPD1, a gene previously linked to spastic paraplegia 64 (Mendelian Inheritance in Man # 615683).
Methods: Individuals with biallelic ENTPD1 variants were recruited worldwide. Deep phenotyping and molecular characterization were performed.
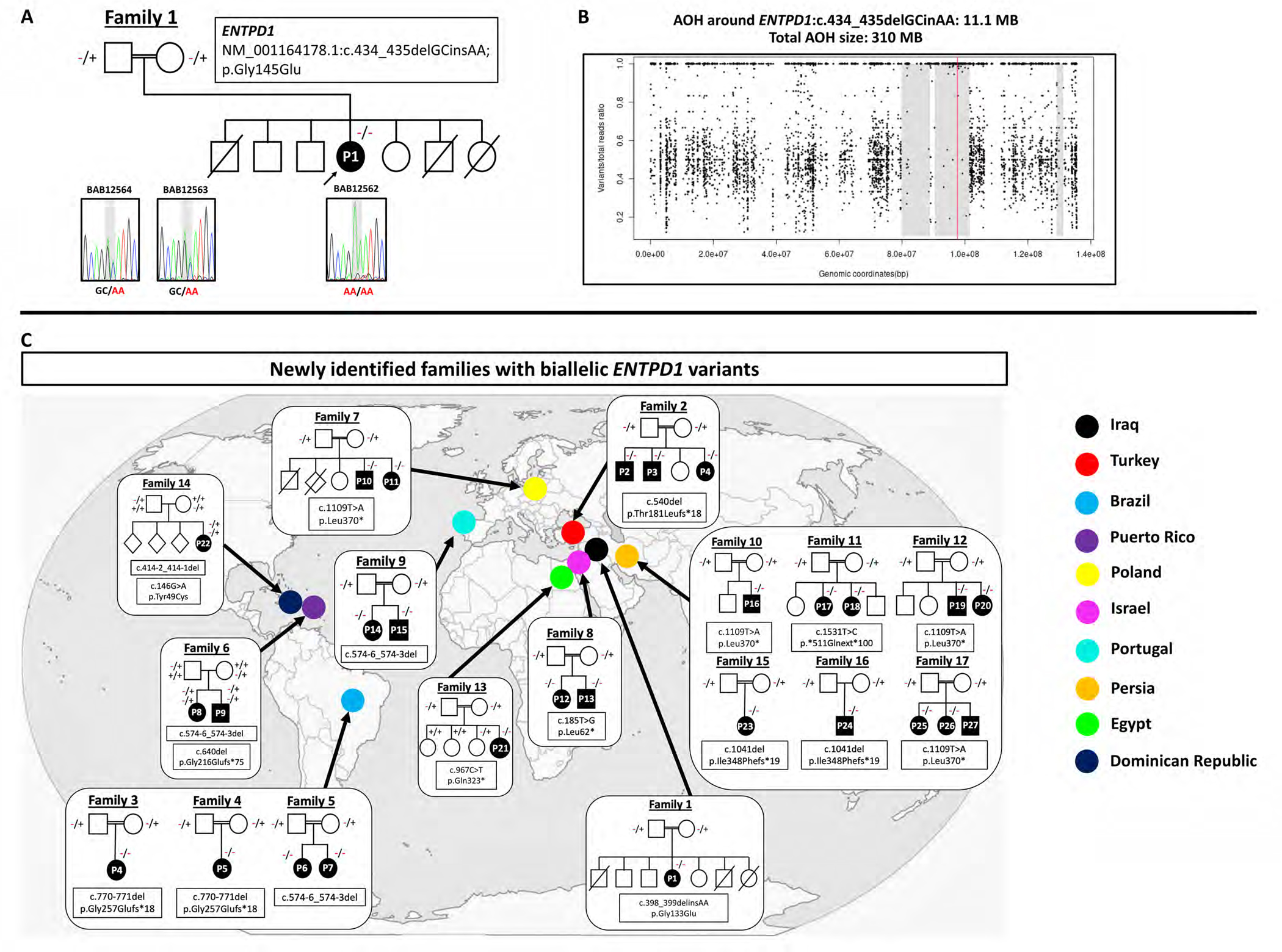
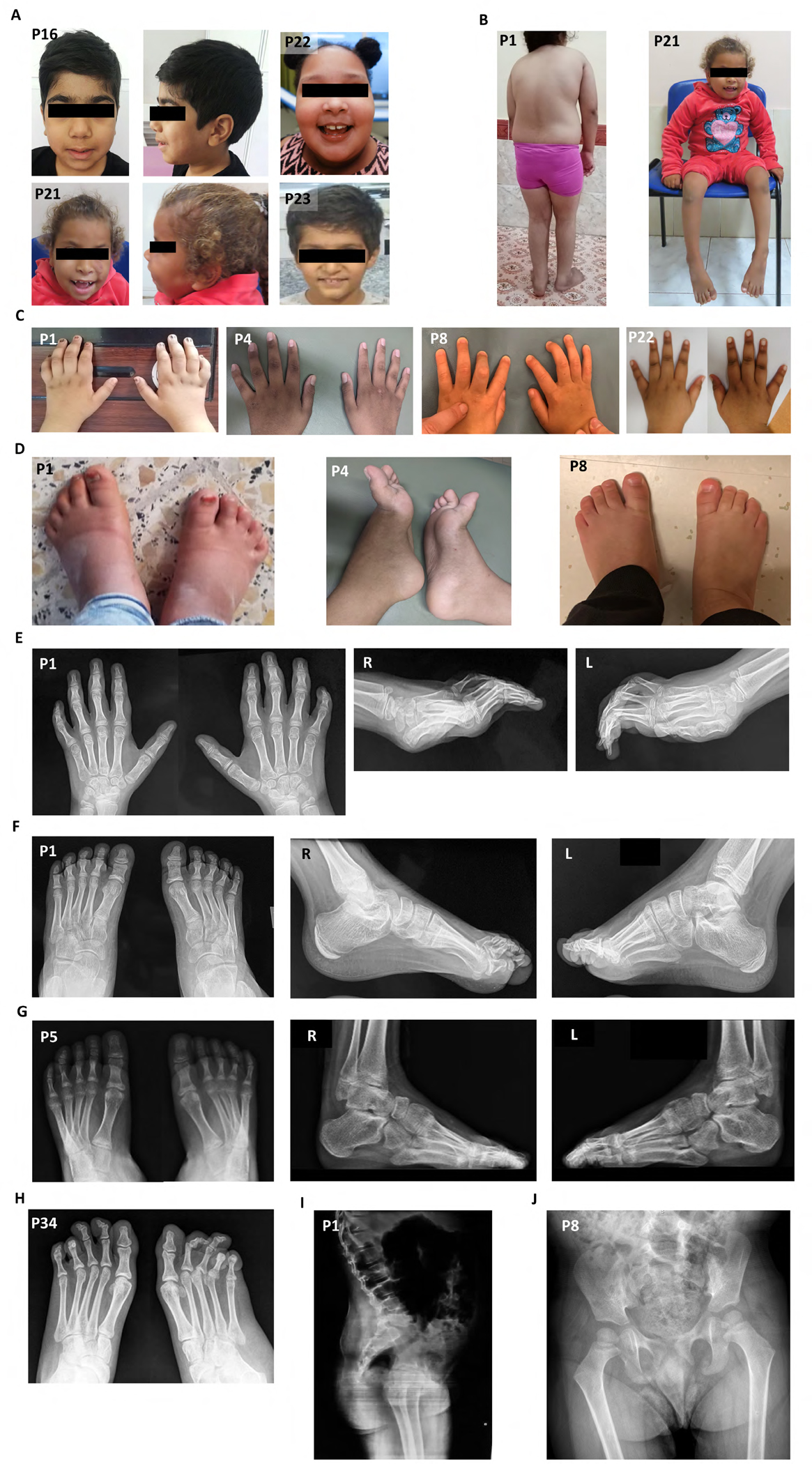
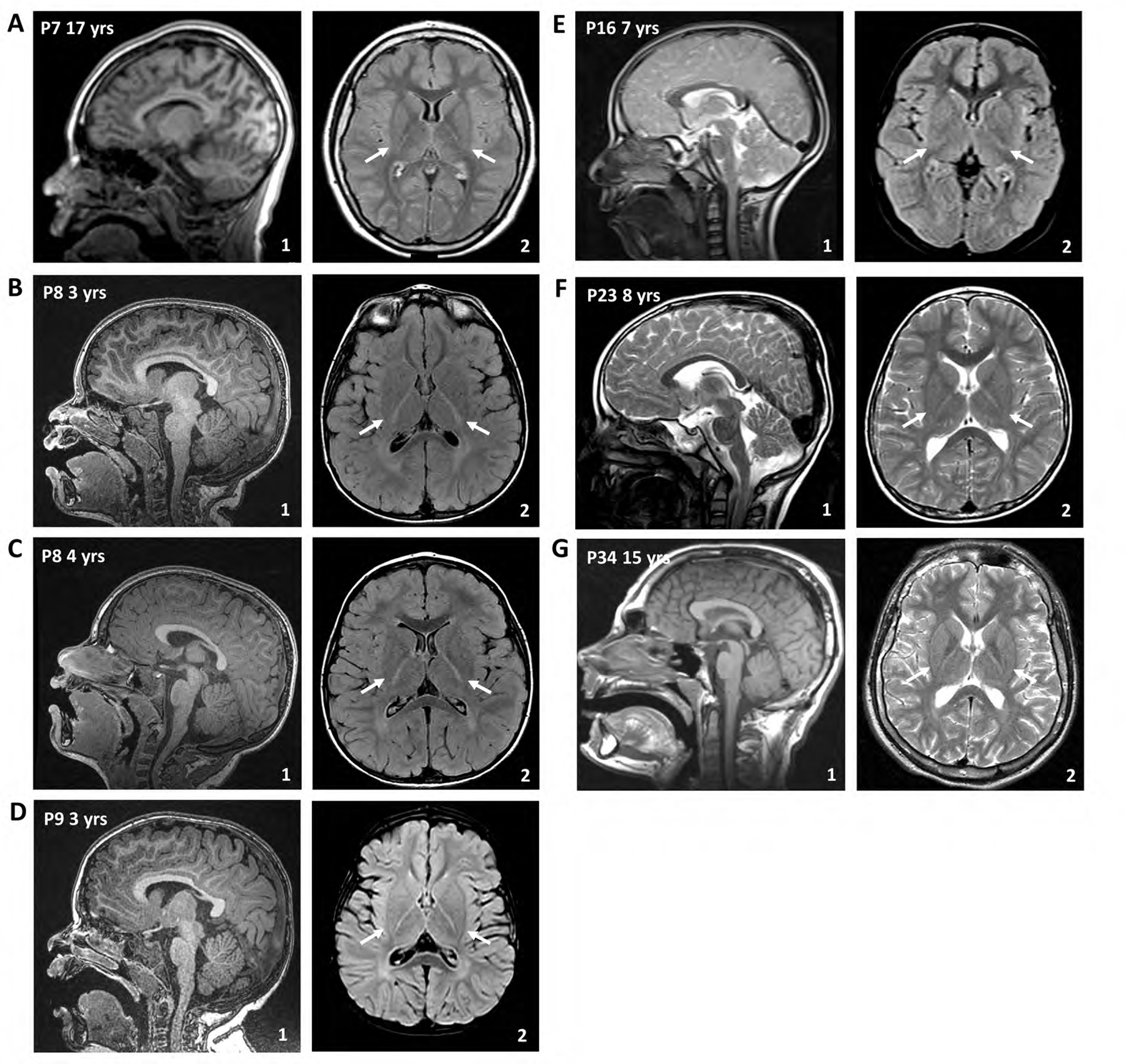
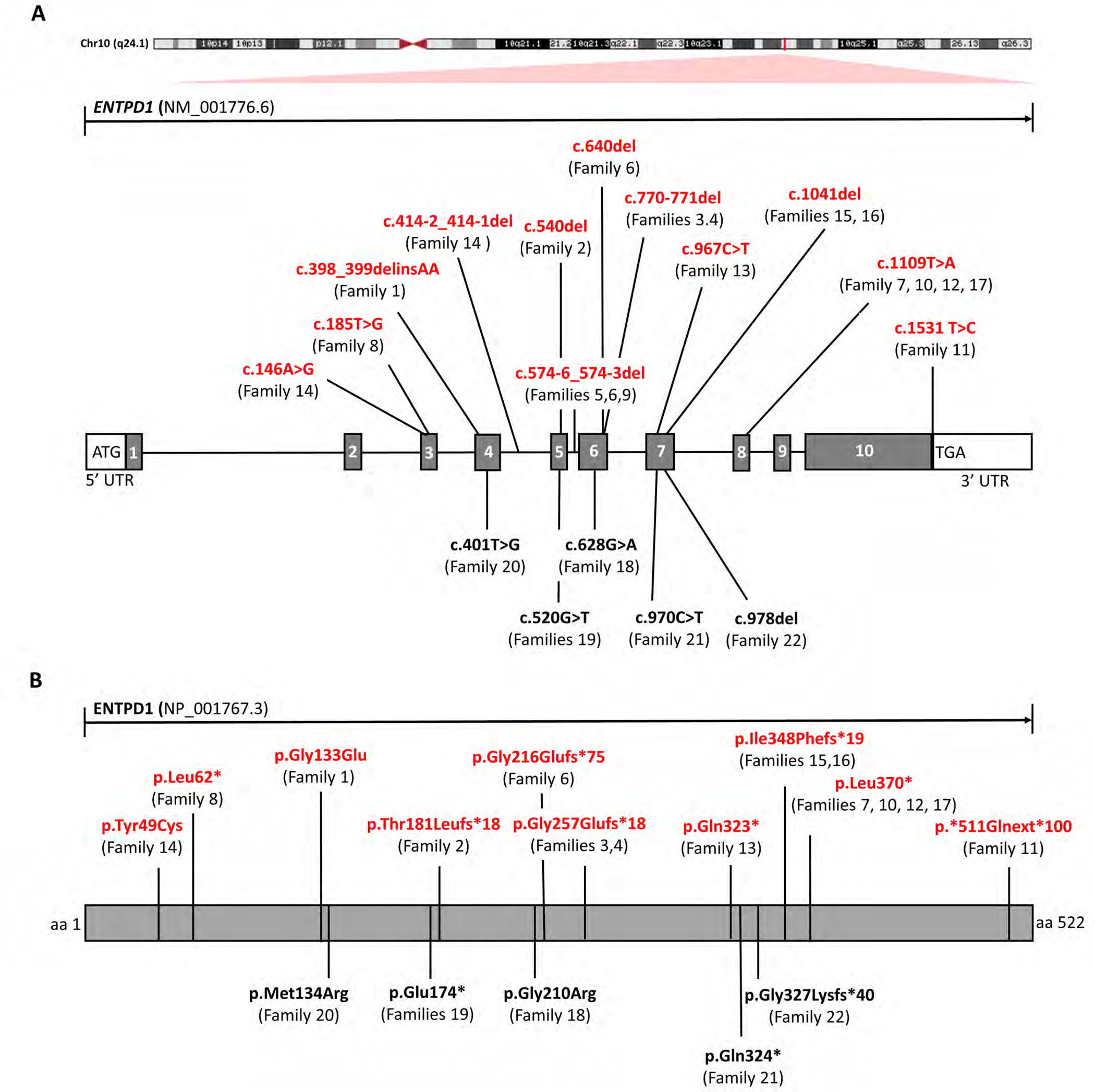
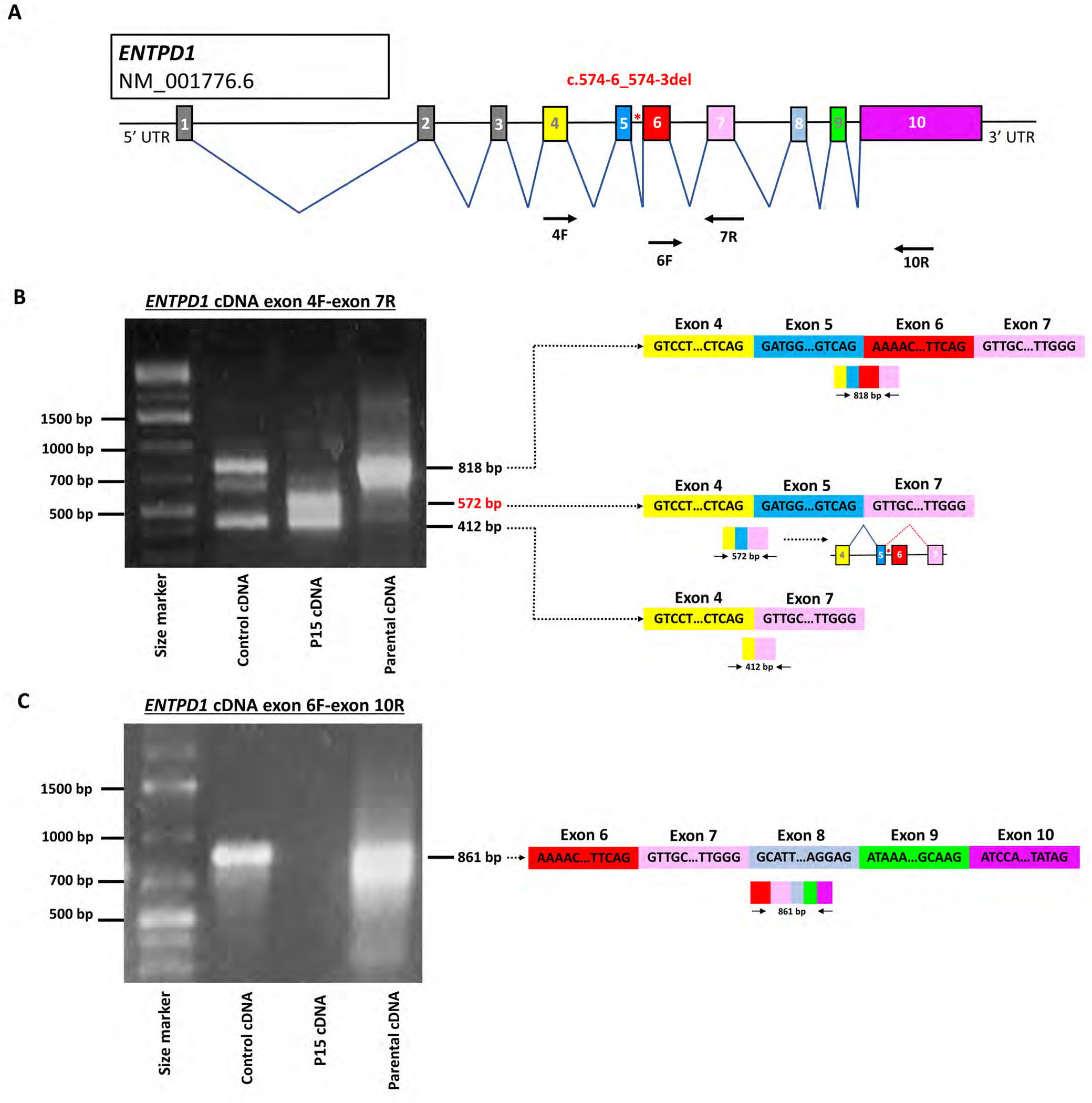
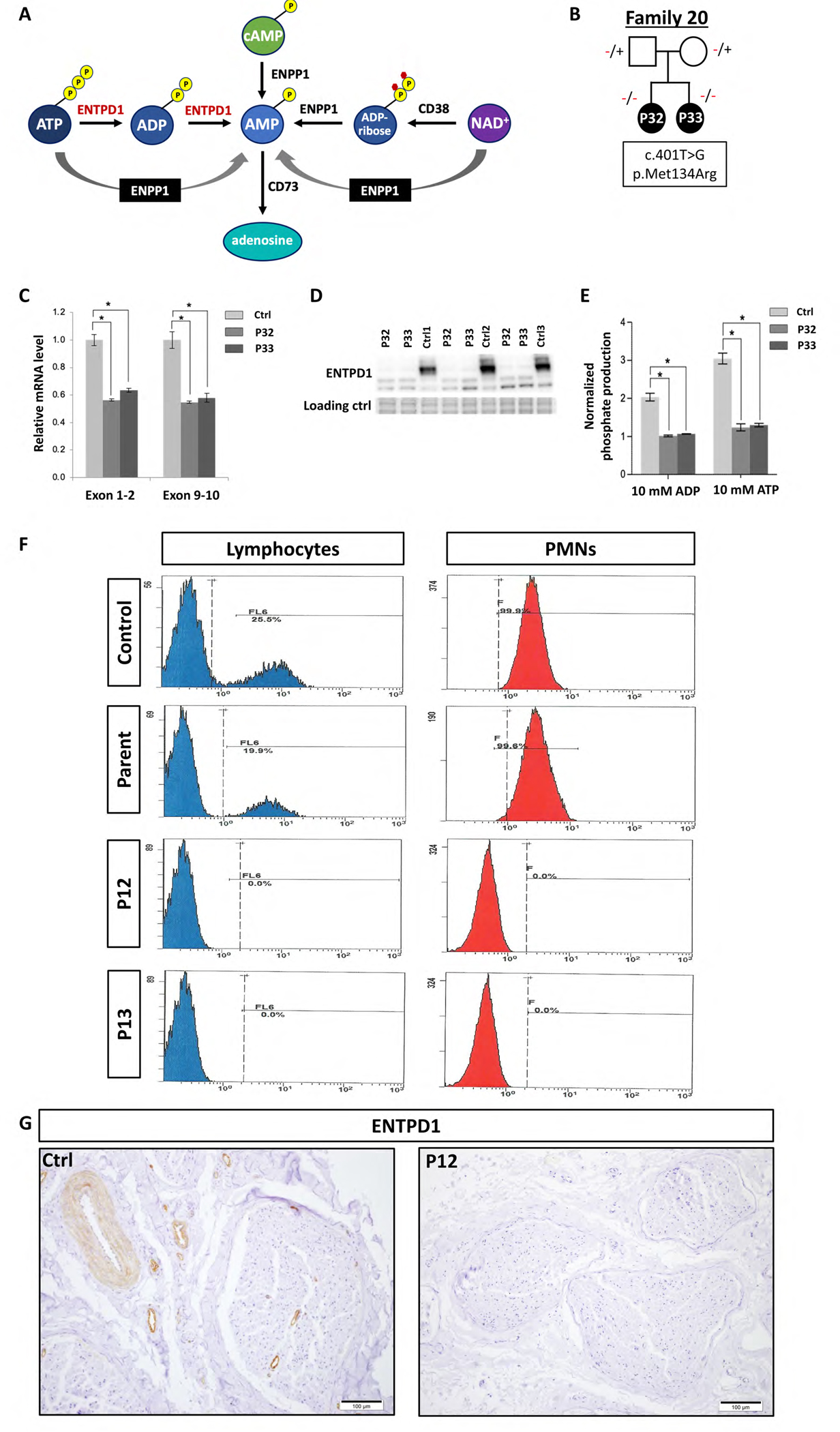
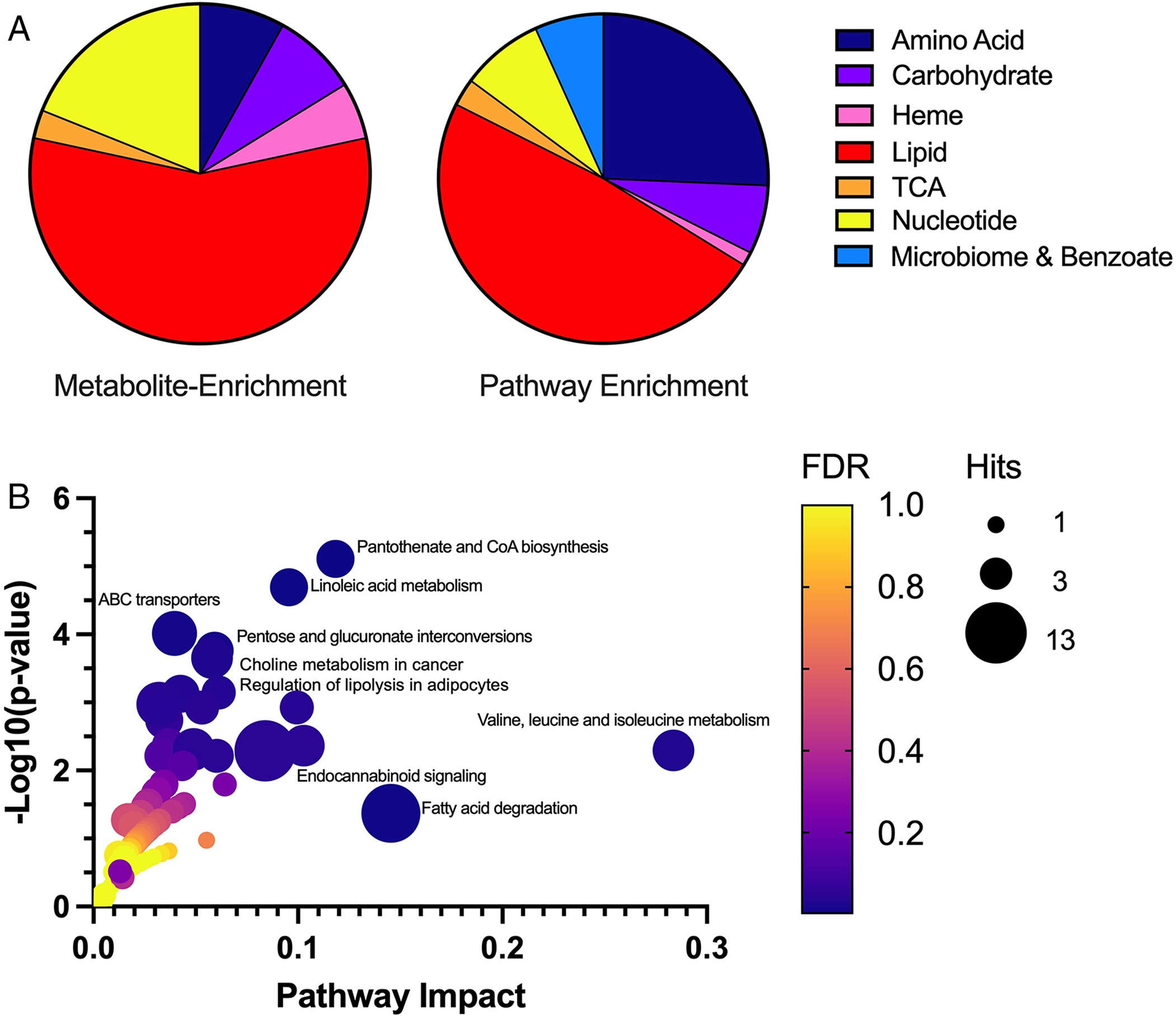
Results: A total of 27 individuals from 17 unrelated families were studied; additional phenotypic information was collected from published cases. Twelve novel pathogenic ENTPD1 variants are described (NM 001776.6): c.398_399delinsAA; p.(Gly133Glu), c.540del; p.(Thr181Leufs*18), c.640del; p.(Gly216Glufs*75), c.185 T > G; p.(Leu62*), c.1531 T > C; p.(*511Glnext*100), c.967C > T; p.(Gln323*), c.414-2_414-1del, and c.146 A > G; p.(Tyr49Cys) including 4 recurrent variants c.1109 T > A; p.(Leu370*), c.574-6_574-3del, c.770_771del; p.(Gly257Glufs*18), and c.1041del; p.(Ile348Phefs*19). Shared disease traits include childhood onset, progressive spastic paraplegia, intellectual disability (ID), dysarthria, and white matter abnormalities. In vitro assays demonstrate that ENTPD1 expression and function are impaired and that c.574-6_574-3del causes exon skipping. Global metabolomics demonstrate ENTPD1 deficiency leads to impaired nucleotide, lipid, and energy metabolism.
Interpretation: The ENTPD1 locus trait consists of childhood disease onset, ID, progressive spastic paraparesis, dysarthria, dysmorphisms, and white matter abnormalities, with some individuals showing neurocognitive regression. Investigation of an allelic series of ENTPD1 (1) expands previously described features of ENTPD1-related neurological disease, (2) highlights the importance of genotype-driven deep phenotyping, (3) documents the need for global collaborative efforts to characterize rare autosomal recessive disease traits, and (4) provides insights into disease trait neurobiology. ANN NEUROL 2022;92:304-321.
© 2022 American Neurological Association.
Conflict of interest statement
Conflicts of interests
J.R.L. has stock ownership in 23andMe, is a paid consultant for Regeneron Genetics Center, and is a co-inventor on multiple United States and European patents related to molecular diagnostics for inherited neuropathies, eye diseases, and bacterial genomic fingerprinting. The Department of Molecular and Human Genetics at Baylor College of Medicine receives revenue from clinical genetic testing conducted at Baylor Genetics (BG) Laboratories. M.C.K. is a paid consultant for PTC Therapeutics and Aeglea. Other authors have no potential conflicts to disclose.
Figures
References
-
- Tadahiro Mitani SI, Gezdirici Alper, Gulec Elif Yilmaz, Punetha Jaya, Fatih Jawid M., Herman Isabella, Gulsen Akay Tayfun Haowei Du, Calame Daniel G., Ayaz Akif, Tos Tulay, Yesil Gozde, Aydin Hatip, Geckinli Bilgen, Elcioglu Nursel, Candan Sukru, Sezer Ozlem, Haktan Bagis Erdem Davut Gul, Yasar Emine, Koparir Erkan, Elmas Muhsin, Yesilbas Osman, Kilic Betul, Serdal Güngör Ahmet C. Ceylan, Bozdogan Sevcan, Ture Mehmet, Etlik Ozdal, Cicek Salih, Aslan Huseyin, Yalcintepe Sinem, Topcu Vehap, Ipek Zeynep, Bayram Yavuz, Grochowski Christopher M., Jolly Angad, Dawood Moez, Duan Ruizhi, Jhangiani Shalini N., Doddapaneni Harsha, Hu Jianhong, Muzny Donna M., Baylor-Hopkins Center for Mendelian Genomics, Marafi Dana, Akdemir Zeynep Coban, Karaca Ender, Carvalho Claudia MBC, Gibbs Richard A., Posey Jennifer E., Lupski James R., and Pehlivan Davut. (2021). Evidence for a higher prevalence of oligogenic inheritance in neurodevelopmental disorders in the Turkish population. American Journal of Human Genetics. - PMC - PubMed
-
- Ruano L, Melo C, Silva MC, and Coutinho P (2014). The global epidemiology of hereditary ataxia and spastic paraplegia: a systematic review of prevalence studies. Neuroepidemiology 42, 174–183. - PubMed
-
- Blackstone C (2018). Hereditary spastic paraplegia. Handb Clin Neurol 148, 633–652. - PubMed
MeSH terms
Substances
Grants and funding
- U54HG003273/HG/NHGRI NIH HHS/United States
- T32 NS043124-19/NH/NIH HHS/United States
- T32 GM007526/GM/NIGMS NIH HHS/United States
- NIH T32 GM007526-42/NH/NIH HHS/United States
- R35 NS105078/NS/NINDS NIH HHS/United States
- T32 NS043124/NS/NINDS NIH HHS/United States
- R01NS106298/NS/NINDS NIH HHS/United States
- R01 NS106298/NS/NINDS NIH HHS/United States
- R21CA164970/NH/NIH HHS/United States
- R35NS105078/NS/NINDS NIH HHS/United States
- U01 HG011758/HG/NHGRI NIH HHS/United States
- MR/S01165X/1/MRC_/Medical Research Council/United Kingdom
- UM1 HG006542/HG/NHGRI NIH HHS/United States
- U54 HG003273/HG/NHGRI NIH HHS/United States
- K08 HG008986/HG/NHGRI NIH HHS/United States
- R01DK108894/NH/NIH HHS/United States
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
