

Direct free energy evaluation of classical and quantum many-body systems via field-theoretic simulation
- PMID: 35471906
- PMCID: PMC9170146
- DOI: 10.1073/pnas.2201804119
Direct free energy evaluation of classical and quantum many-body systems via field-theoretic simulation
Abstract
Free energy evaluation in molecular simulations of both classical and quantum systems is computationally intensive and requires sophisticated algorithms. This is because free energy depends on the volume of accessible phase space, a quantity that is inextricably linked to the integration measure in a coordinate representation of a many-body problem. In contrast, the same problem expressed as a field theory (auxiliary field or coherent states) isolates the particle number as a simple parameter in the Hamiltonian or action functional and enables the identification of a chemical potential field operator. We show that this feature leads a “direct” method of free energy evaluation, in which a particle model is converted to a field theory and appropriate field operators are averaged using a field-theoretic simulation conducted with complex Langevin sampling. These averages provide an immediate estimate of the Helmholtz free energy in the canonical ensemble and the entropy in the microcanonical ensemble. The method is illustrated for a classical polymer solution, a block copolymer melt exhibiting liquid crystalline and solid mesophases, and a quantum fluid of interacting bosons.
Keywords: field-theoretic simulation; free energy; molecular simulation; polymers; quantum fluids.
Conflict of interest statement
The authors declare no competing interest.
Figures

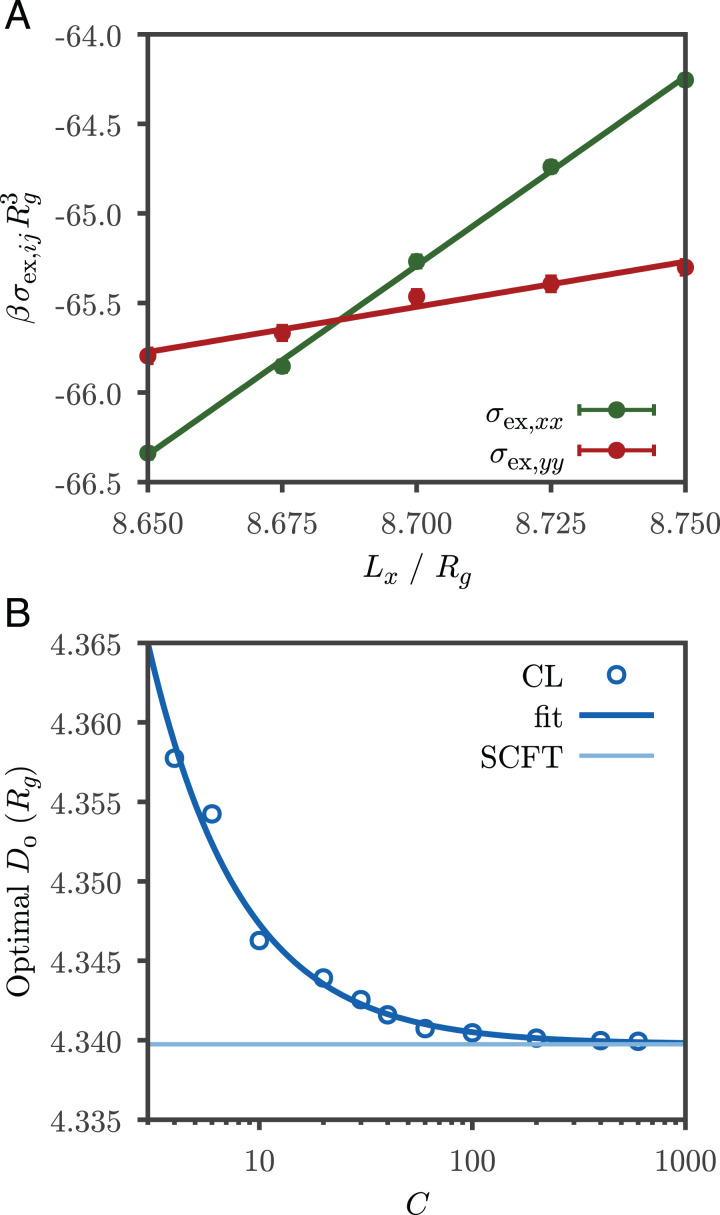


References
-
- Frenkel D., Smit B., Understanding Molecular Simulation (Academic Press, London, United Kingdom, 1996).
-
- Allen M. P., Tildesley D. J., Computer Simulation of Liquids (Oxford University Press, New York, NY, 1987).
-
- Landau D. P., Binder K., A Guide to Monte Carlo Simulation in Statistical Physics (Cambridge University Press, Cambridge, United Kingdom, 2000).
-
- Shell M. S., Panagiotopoulos A., Pohorille A., “Methods based on probability distributions and histograms” in Springer Series in Chemical Physics, Chipot C., Pohorille A., Eds. (Springer, Berlin, Germany, 2007), vol. 86, pp. 77–118.
-
- Widom B., Some topics in the theory of fluids. J. Chem. Phys. 39, 2808–2812 (1963).
