

Multi-targeting of K-Ras domains and mutations by peptide and small molecule inhibitors
- PMID: 35472201
- PMCID: PMC9041843
- DOI: 10.1371/journal.pcbi.1009962
Multi-targeting of K-Ras domains and mutations by peptide and small molecule inhibitors
Abstract
K-Ras activating mutations are significantly associated with tumor progression and aggressive metastatic behavior in various human cancers including pancreatic cancer. So far, despite a large number of concerted efforts, targeting of mutant-type K-Ras has not been successful. In this regard, we aimed to target this oncogene by a combinational approach consisting of small peptide and small molecule inhibitors. Based on a comprehensive analysis of structural and physicochemical properties of predominantly K-Ras mutants, an anti-cancer peptide library and a small molecule library were screened to simultaneously target oncogenic mutations and functional domains of mutant-type K-Ras located in the P-loop, switch I, and switch II regions. The selected peptide and small molecule showed notable binding affinities to their corresponding binding sites, and hindered the growth of tumor cells carrying K-RasG12D and K-RasG12C mutations. Of note, the expression of K-Ras downstream genes (i.e., CTNNB1, CCND1) was diminished in the treated Kras-positive cells. In conclusion, our combinational platform signifies a new potential for blockade of oncogenic K-Ras and thereby prevention of tumor progression and metastasis. However, further validations are still required regarding the in vitro and in vivo efficacy and safety of this approach.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
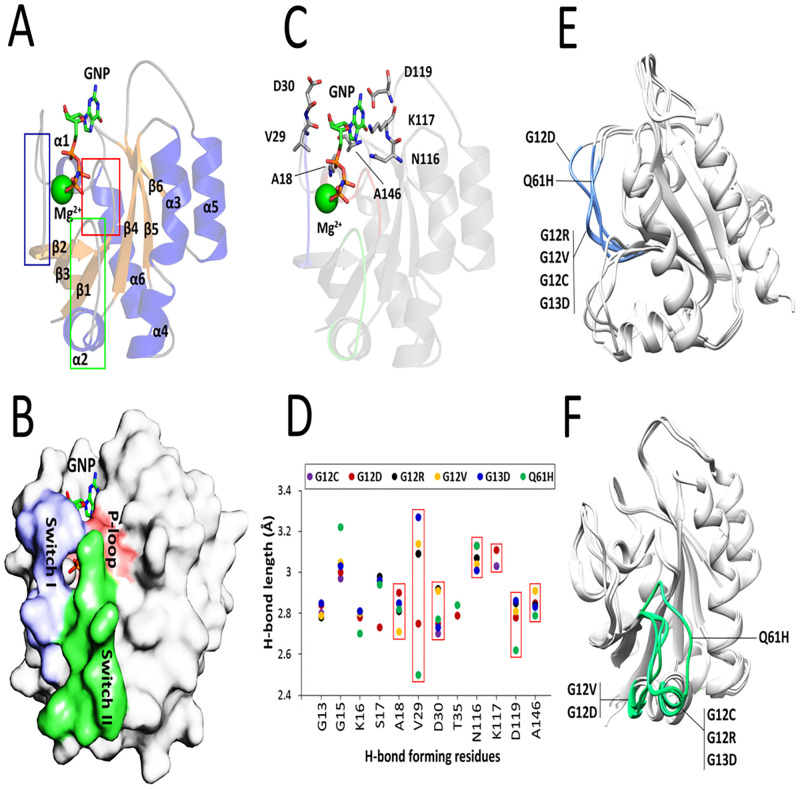
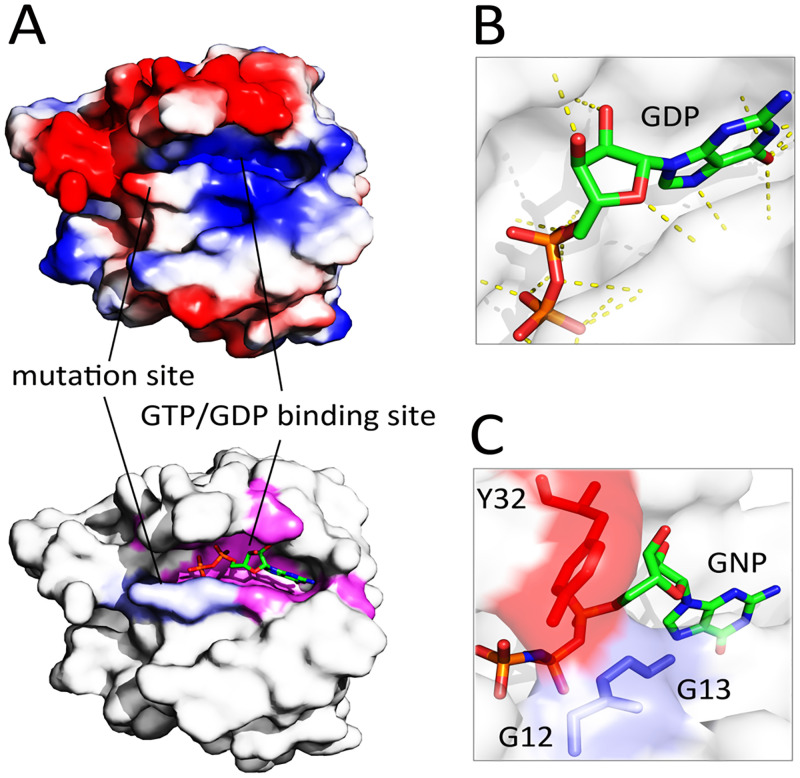
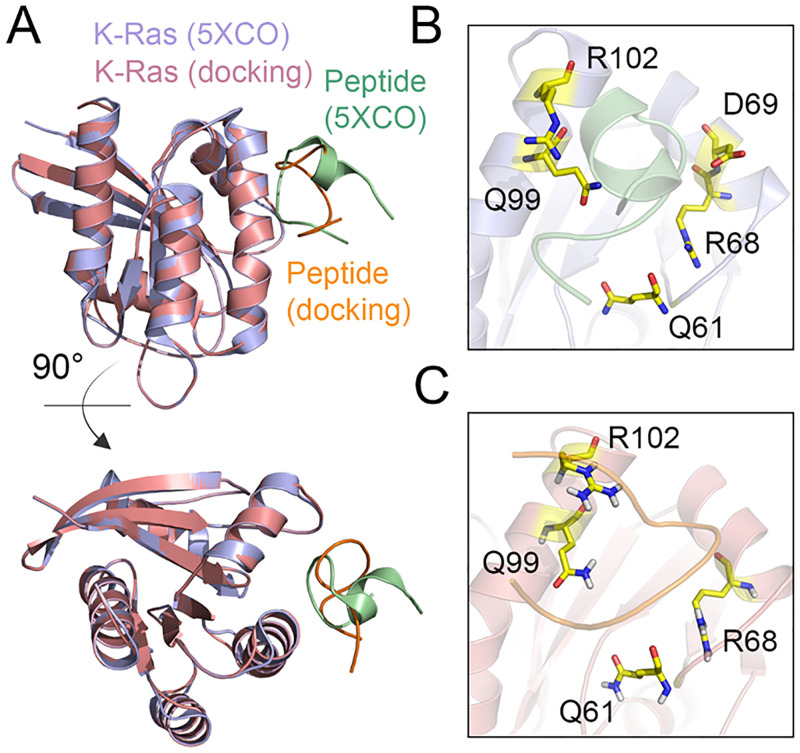
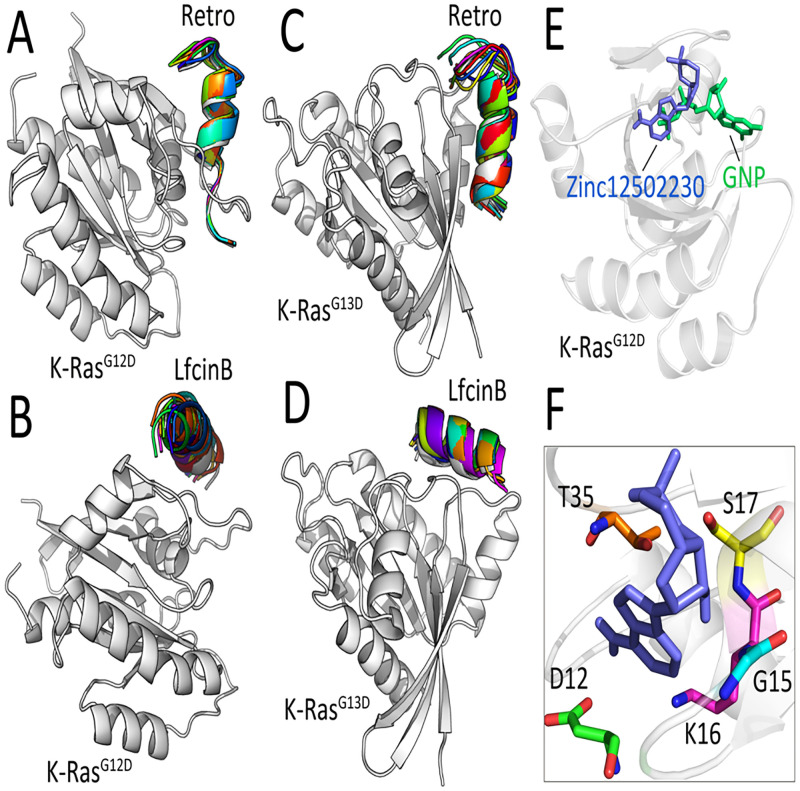
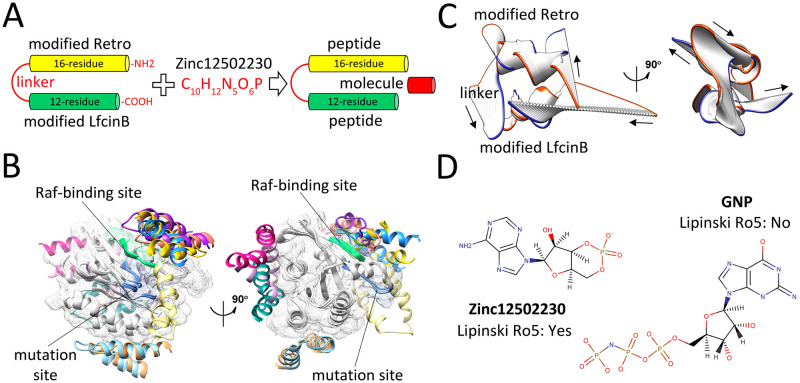
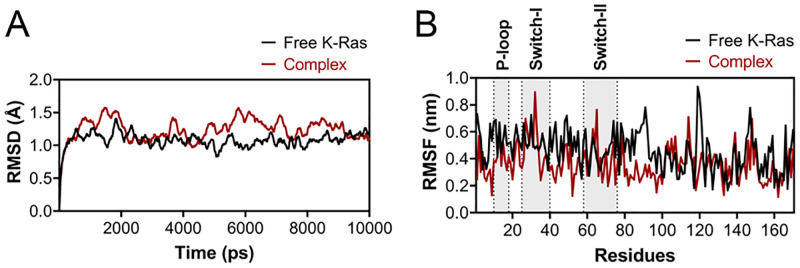
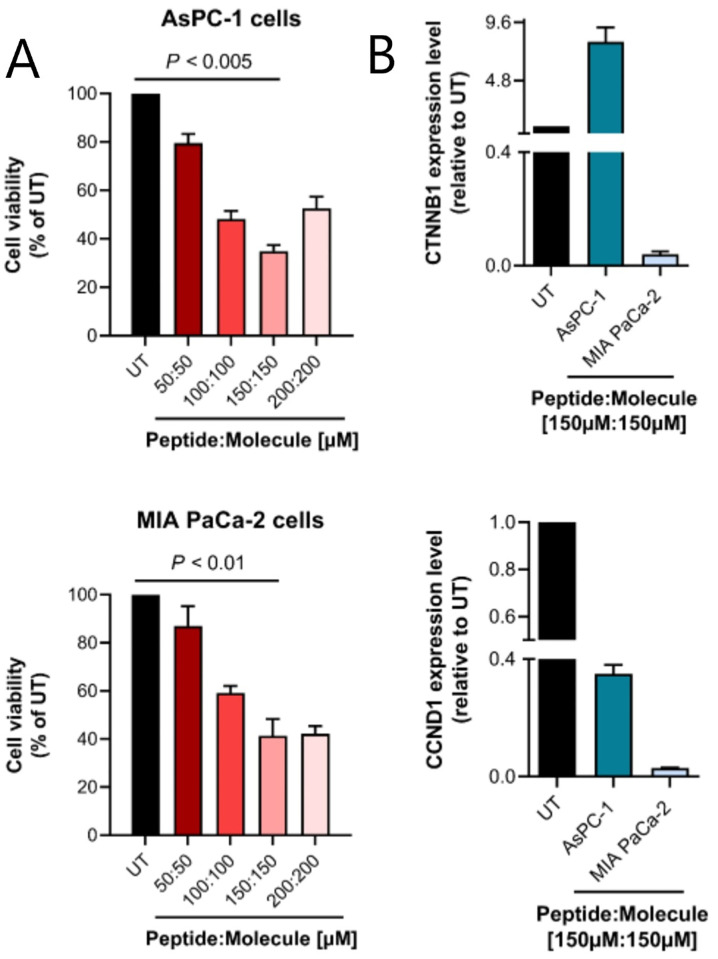
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
