

Systematic discovery and validation of T cell targets directed against oncogenic KRAS mutations
- PMID: 35474673
- PMCID: PMC9017224
- DOI: 10.1016/j.crmeth.2021.100084
Systematic discovery and validation of T cell targets directed against oncogenic KRAS mutations
Abstract
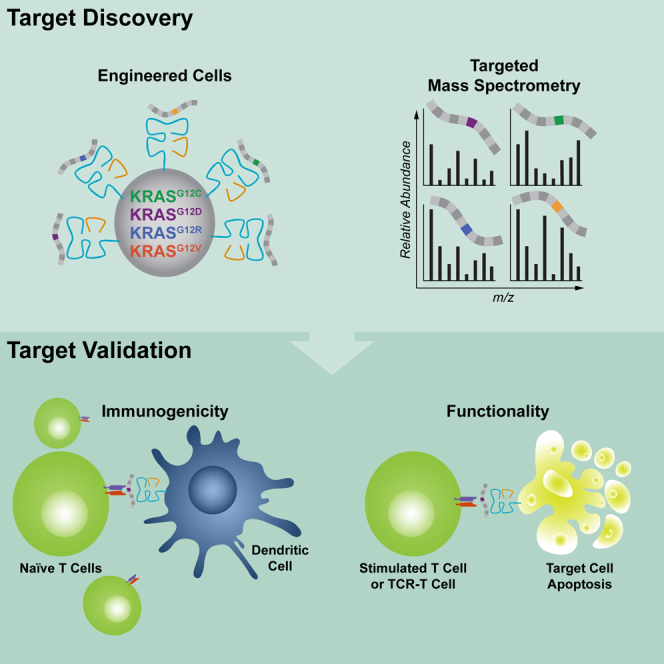
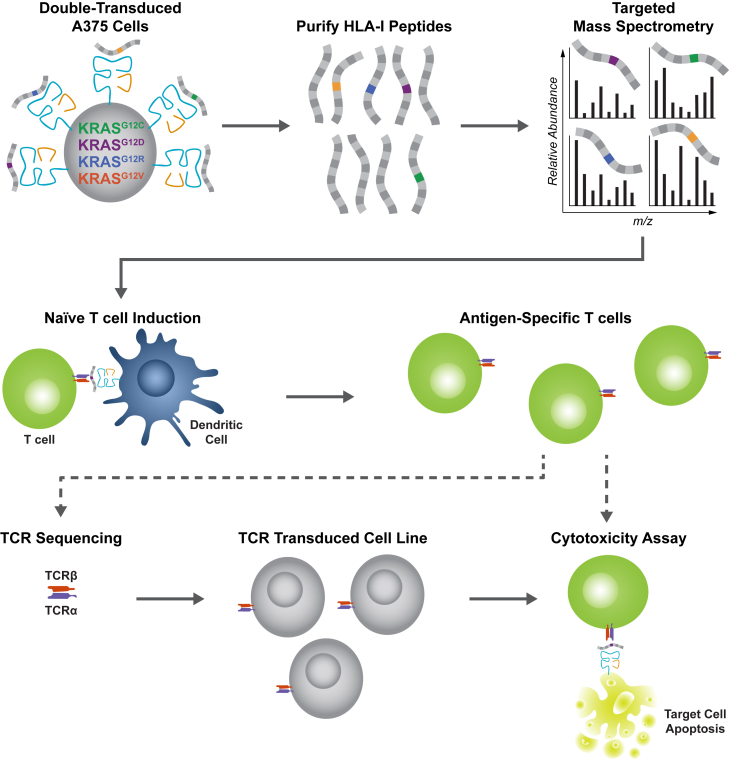
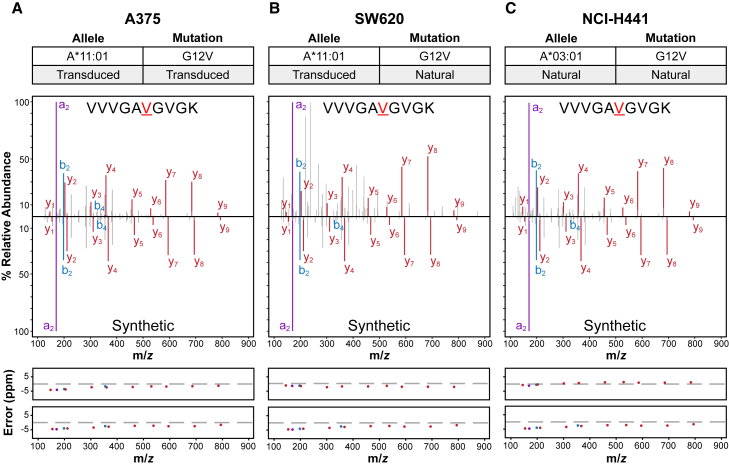
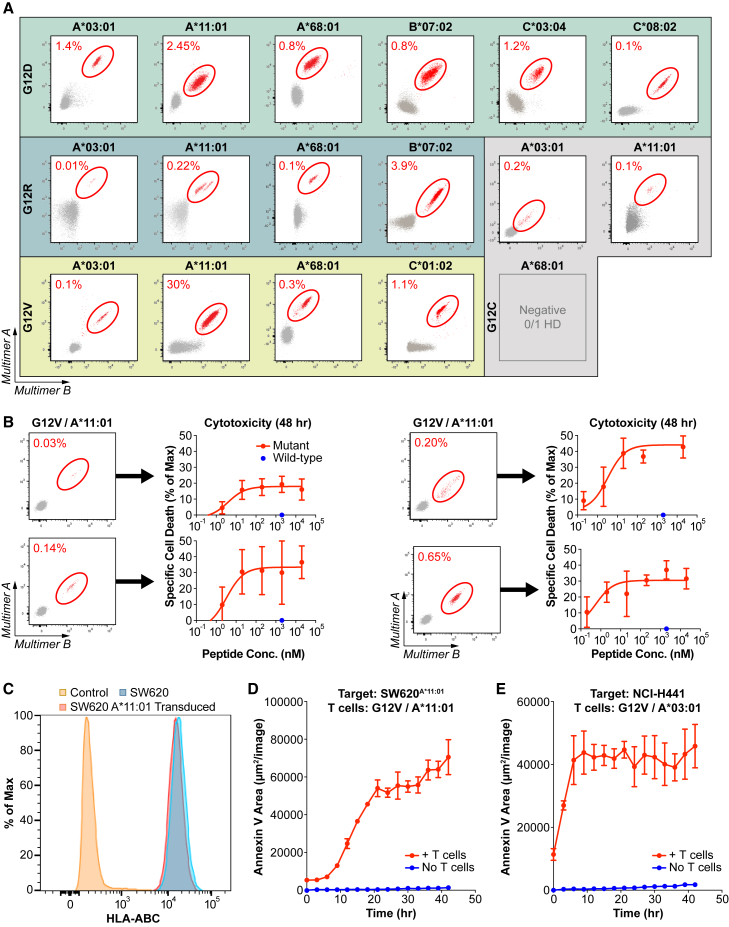
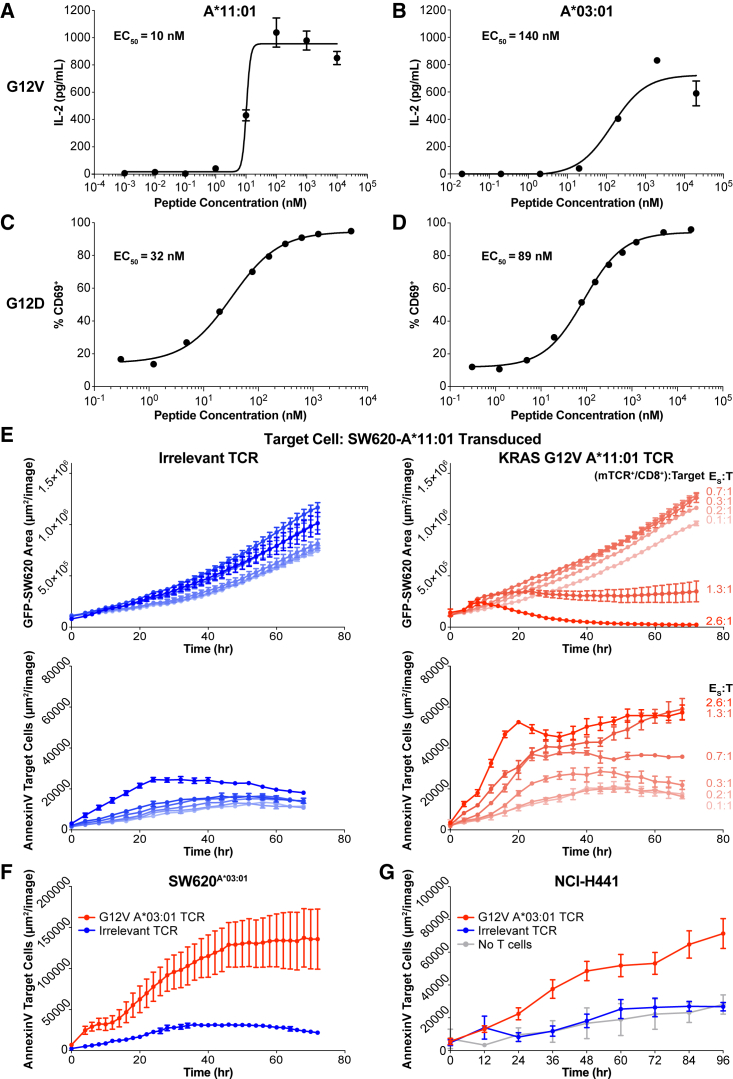
Oncogenic mutations in KRAS can be recognized by T cells on specific class I human leukocyte antigen (HLA-I) molecules, leading to tumor control. To date, the discovery of T cell targets from KRAS mutations has relied on occasional T cell responses in patient samples or the use of transgenic mice. To overcome these limitations, we have developed a systematic target discovery and validation pipeline. We evaluate the presentation of mutant KRAS peptides on individual HLA-I molecules using targeted mass spectrometry and identify 13 unpublished KRASG12C/D/R/V mutation/HLA-I pairs and nine previously described pairs. We assess immunogenicity, generating T cell responses to nearly all targets. Using cytotoxicity assays, we demonstrate that KRAS-specific T cells and T cell receptors specifically recognize endogenous KRAS mutations. The discovery and validation of T cell targets from KRAS mutations demonstrate the potential for this pipeline to aid the development of immunotherapies for important cancer targets.
Keywords: HLA; PRM; RAS; T cell; TCR; cancer; immunotherapy; mass spectrometry; neoantigen.
© 2021 The Authors.
Conflict of interest statement
J.C., S.P.G., B.P.C., J.L.D., J.K., D.L., D.A.R., P.J.T., J.R.S., K.C.F., M.S.R., M.M.v.B., R.B.G., T.A.A., and V.R.J. are employees and shareholders of BioNTech SE. C.D.McG., A.B., and J.G.A. are former employees of Neon Therapeutics (acquired by BioNTech SE). C.D.McG. is a graduate student at the University of Washington. A.B. is an employee and shareholder of TScan Therapeutics. J.G.A. is an employee of the Broad Institute of MIT and Harvard. R.B.G. is a member of the board of directors at Alkermes plc and Infinity Pharmaceuticals, and a member of the scientific advisory board at Leap Therapeutics.
Figures
References
-
- Abelin J.G., Harjanto D., Malloy M., Suri P., Colson T., Goulding S.P., Creech A.L., Serrano L.R., Nasir G., Nasrullah Y., et al. Defining HLA-II ligand processing and binding rules with mass spectrometry enhances cancer epitope prediction. Immunity. 2019;51:766–779.e17. - PubMed
-
- Ali M., Foldvari Z., Giannakopoulou E., Böschen M.-L., Strønen E., Yang W., Toebes M., Schubert B., Kohlbacher O., Schumacher T.N., et al. Induction of neoantigen-reactive T cells from healthy donors. Nat. Protoc. 2019;14:1926–1943. - PubMed
-
- Bar-Sagi D., Knelson E.H., Sequist L.V. A bright future for KRAS inhibitors. Nat. Cancer. 2020;1:25–27. - PubMed
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
