

Super-resolved live-cell imaging using random illumination microscopy
- PMID: 35474693
- PMCID: PMC9017237
- DOI: 10.1016/j.crmeth.2021.100009
Super-resolved live-cell imaging using random illumination microscopy
Abstract
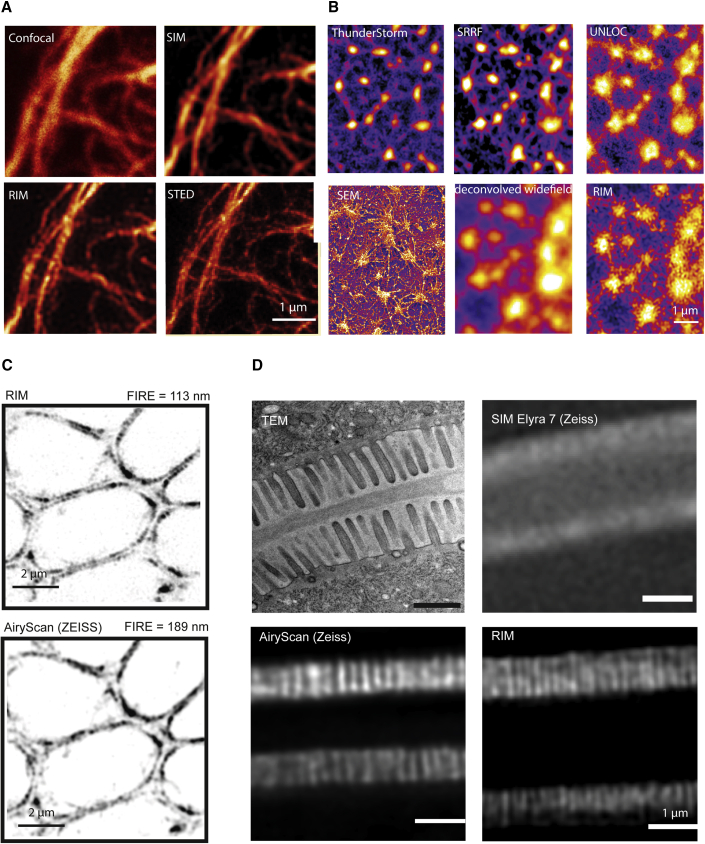
Current super-resolution microscopy (SRM) methods suffer from an intrinsic complexity that might curtail their routine use in cell biology. We describe here random illumination microscopy (RIM) for live-cell imaging at super-resolutions matching that of 3D structured illumination microscopy, in a robust fashion. Based on speckled illumination and statistical image reconstruction, easy to implement and user-friendly, RIM is unaffected by optical aberrations on the excitation side, linear to brightness, and compatible with multicolor live-cell imaging over extended periods of time. We illustrate the potential of RIM on diverse biological applications, from the mobility of proliferating cell nuclear antigen (PCNA) in U2OS cells and kinetochore dynamics in mitotic S. pombe cells to the 3D motion of myosin minifilaments deep inside Drosophila tissues. RIM's inherent simplicity and extended biological applicability, particularly for imaging at increased depths, could help make SRM accessible to biology laboratories.
Keywords: aberration; computational imaging; fluorescence; live imaging; microscopy; speckle; super-resolution; variance.
© 2021 The Authors.
Conflict of interest statement
The authors declare no competing interests. AlgoRIM described herein has been filed by S.L., T.M., J.I., M.A., and A.S. on April 30, 2020, at the Agence de Protection des Programmes under the number: IDDN.FR.001.180008.000.S.P.2020.000.00000.
Figures





References
-
- Ayuk R., Giovannini H., Jost A., Mudry E., Girard J., Mangeat T., Sandeau N., Heintzmann R., Wicker K., Belkebir K., et al. Structured illumination fluorescence microscopy with distorted excitations using a filtered blind-SIM algorithm. Opt. Lett. 2013;38:4723–4726. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Molecular Biology Databases
Miscellaneous
