

A comprehensive approach for genome-wide efficiency profiling of DNA modifying enzymes
- PMID: 35475220
- PMCID: PMC9017147
- DOI: 10.1016/j.crmeth.2022.100187
A comprehensive approach for genome-wide efficiency profiling of DNA modifying enzymes
Abstract
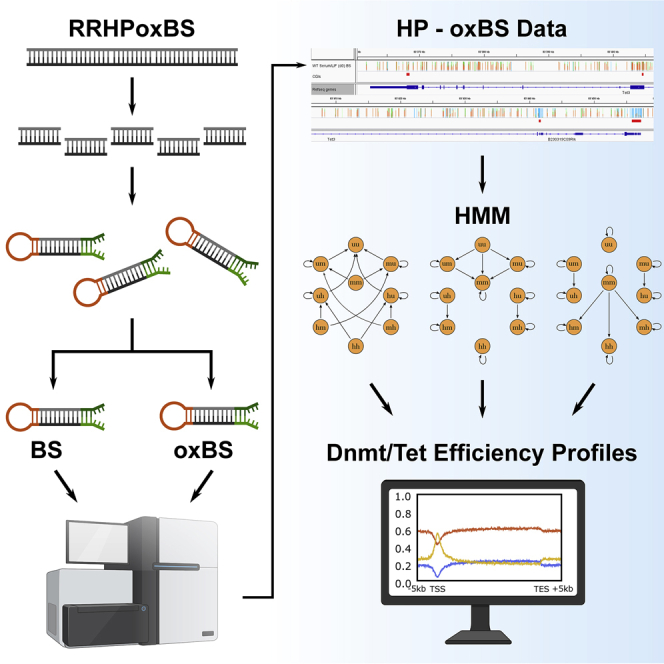
A precise understanding of DNA methylation dynamics is of great importance for a variety of biological processes including cellular reprogramming and differentiation. To date, complex integration of multiple and distinct genome-wide datasets is required to realize this task. We present GwEEP (genome-wide epigenetic efficiency profiling) a versatile approach to infer dynamic efficiencies of DNA modifying enzymes. GwEEP relies on genome-wide hairpin datasets, which are translated by a hidden Markov model into quantitative enzyme efficiencies with reported confidence around the estimates. GwEEP predicts de novo and maintenance methylation efficiencies of Dnmts and furthermore the hydroxylation efficiency of Tets. Its design also allows capturing further oxidation processes given available data. We show that GwEEP predicts accurately the epigenetic changes of ESCs following a Serum-to-2i shift and applied to Tet TKO cells confirms the hypothesized mutual interference between Dnmts and Tets.
Keywords: 5hmC; 5mC; DNA demethylation; DNA methylation; Dnmts; Tets; epigenetics; hairpin sequencing; hidden Markov model.
© 2022 The Authors.
Conflict of interest statement
W.R. is a consultant and shareholder of Cambridge Epigenetix. All other authors declare no competing interests.
Figures





References
-
- Arita K., Ariyoshi M., Tochio H., Nakamura Y., Shirakawa M. Recognition of hemi-methylated dna by the sra protein uhrf1 by a base-flipping mechanism. Nature. 2008;455:818–821. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
