

PeakVI: A deep generative model for single-cell chromatin accessibility analysis
- PMID: 35475224
- PMCID: PMC9017241
- DOI: 10.1016/j.crmeth.2022.100182
PeakVI: A deep generative model for single-cell chromatin accessibility analysis
Abstract
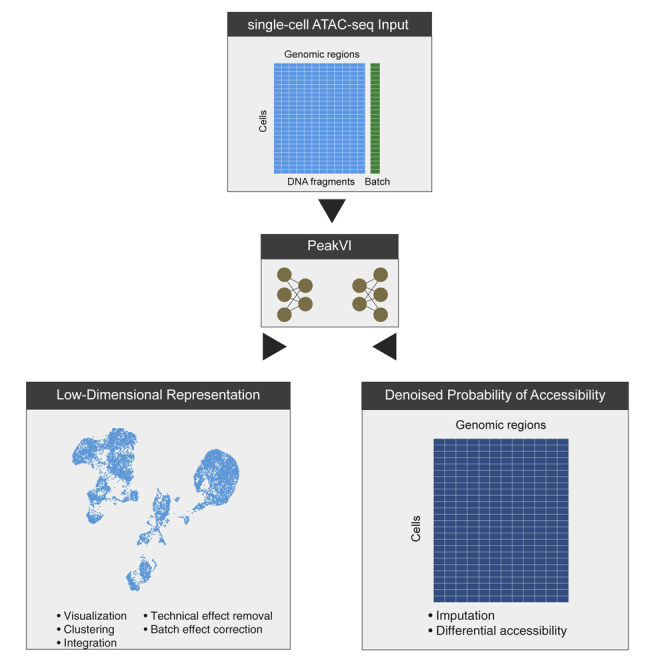
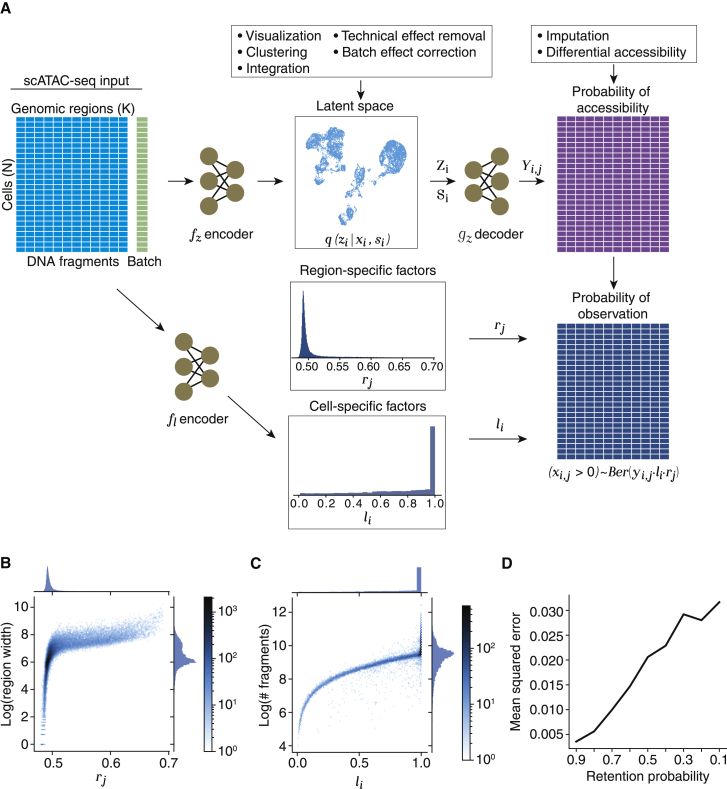
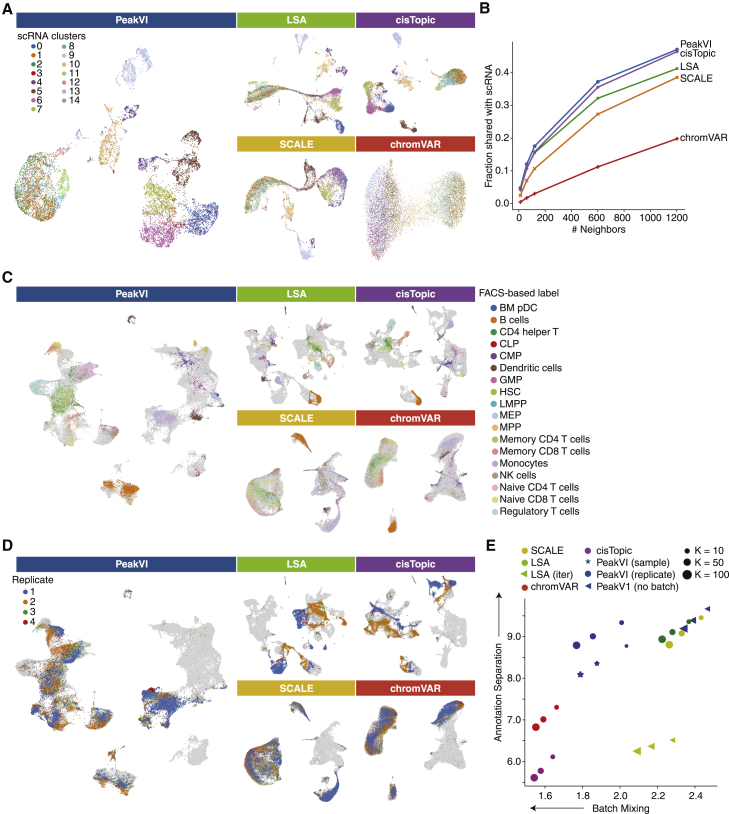
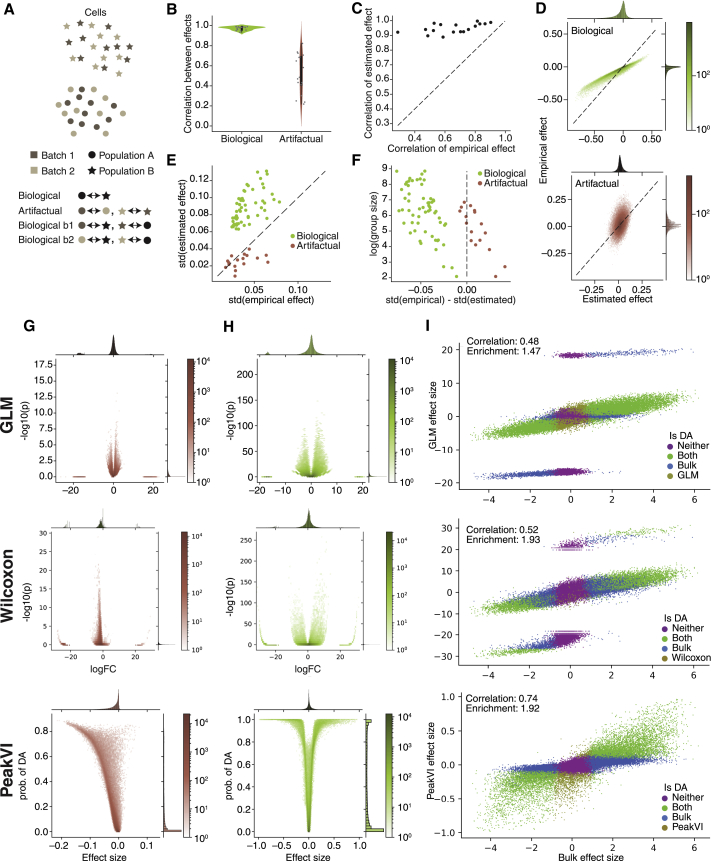
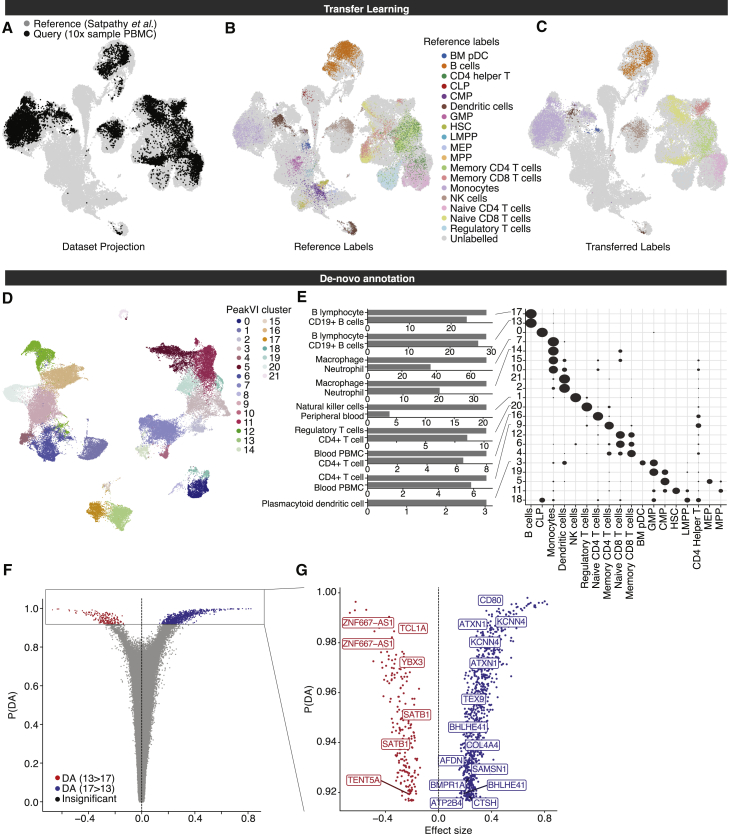
Single-cell ATAC sequencing (scATAC-seq) is a powerful and increasingly popular technique to explore the regulatory landscape of heterogeneous cellular populations. However, the high noise levels, degree of sparsity, and scale of the generated data make its analysis challenging. Here, we present PeakVI, a probabilistic framework that leverages deep neural networks to analyze scATAC-seq data. PeakVI fits an informative latent space that preserves biological heterogeneity while correcting batch effects and accounting for technical effects, such as library size and region-specific biases. In addition, PeakVI provides a technique for identifying differential accessibility at a single-region resolution, which can be used for cell-type annotation as well as identification of key cis-regulatory elements. We use public datasets to demonstrate that PeakVI is scalable, stable, robust to low-quality data, and outperforms current analysis methods on a range of critical analysis tasks. PeakVI is publicly available and implemented in the scvi-tools framework.
Keywords: deep learning; single-cell ATAC-seq; single-cell chromatin accessibility; single-cell genomics.
© 2022 The Authors.
Conflict of interest statement
All authors declare no competing interests.
Figures
References
-
- Carlson M., Maintainer B.P. 2015. TxDb.Hsapiens.UCSC.hg19.knownGene: Annotation package for TxDb object(s). (R package version 3.2.2.) https://bioconductor.org/packages/release/data/annotation/html/TxDb.Hsap....
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
