

ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs
- PMID: 35477154
- PMCID: PMC9157047
- DOI: 10.1038/s41586-022-04769-z
ABO genotype alters the gut microbiota by regulating GalNAc levels in pigs
Abstract
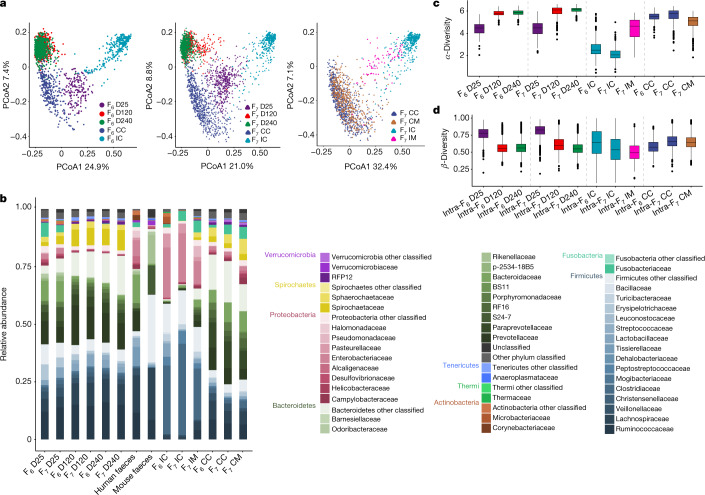
The composition of the intestinal microbiome varies considerably between individuals and is correlated with health1. Understanding the extent to which, and how, host genetics contributes to this variation is essential yet has proved to be difficult, as few associations have been replicated, particularly in humans2. Here we study the effect of host genotype on the composition of the intestinal microbiota in a large mosaic pig population. We show that, under conditions of exacerbated genetic diversity and environmental uniformity, microbiota composition and the abundance of specific taxa are heritable. We map a quantitative trait locus affecting the abundance of Erysipelotrichaceae species and show that it is caused by a 2.3 kb deletion in the gene encoding N-acetyl-galactosaminyl-transferase that underpins the ABO blood group in humans. We show that this deletion is a ≥3.5-million-year-old trans-species polymorphism under balancing selection. We demonstrate that it decreases the concentrations of N-acetyl-galactosamine in the gut, and thereby reduces the abundance of Erysipelotrichaceae that can import and catabolize N-acetyl-galactosamine. Our results provide very strong evidence for an effect of the host genotype on the abundance of specific bacteria in the intestine combined with insights into the molecular mechanisms that underpin this association. Our data pave the way towards identifying the same effect in rural human populations.
© 2022. The Author(s), under exclusive licence to Springer Nature Limited.
Conflict of interest statement
The authors declare no competing interests.
Figures

















Comment in
-
Blood group-gut microbiome-health axis gains further support from landmark multi-omics study in swines.Sci China Life Sci. 2022 Nov;65(11):2338-2340. doi: 10.1007/s11427-022-2145-1. Epub 2022 Aug 5. Sci China Life Sci. 2022. PMID: 35943689 Free PMC article. No abstract available.
References
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
