

The cytotoxic action of BCI is not dependent on its stated DUSP1 or DUSP6 targets in neuroblastoma cells
- PMID: 35478300
- PMCID: PMC9249316
- DOI: 10.1002/2211-5463.13418
The cytotoxic action of BCI is not dependent on its stated DUSP1 or DUSP6 targets in neuroblastoma cells
Abstract
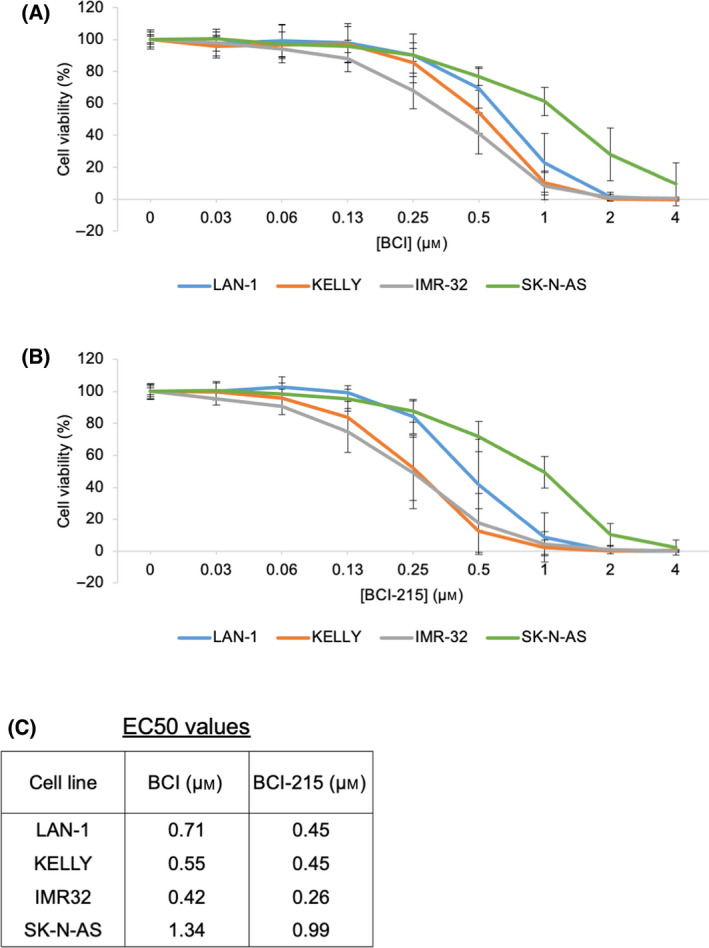
Neuroblastoma (NB) is a heterogeneous cancer of the sympathetic nervous system, which accounts for 7-10% of paediatric malignancies worldwide. Due to the lack of targetable molecular aberrations in NB, most treatment options remain relatively nonspecific. Here, we investigated the therapeutic potential of BCI, an inhibitor of DUSP1 and DUSP6, in cultured NB cells. BCI was cytotoxic in a range of NB cell lines and induced a short-lived activation of the AKT and stress-inducible MAP kinases, although ERK phosphorylation was unaffected. Furthermore, a phosphoproteomic screen identified significant upregulation of JNK signalling components and suppression in mTOR and R6K signalling. To assess the specificity of BCI, CRISPR-Cas9 was employed to introduce insertions and deletions in the DUSP1 and DUSP6 genes. Surprisingly, BCI remained fully cytotoxic in NB cells with complete loss of DUSP6 and partial depletion of DUSP1, suggesting that BCI exerts cytotoxicity in NB cells through a complex mechanism that is unrelated to these phosphatases. Overall, these data highlight the risk of using an inhibitor such as BCI as supposedly specific DUSP1/6, without understanding its full range of targets in cancer cells.
Keywords: CRISPR-Cas9; MAPK signalling; chemical inhibition; dual-specificity phosphatase; neuroblastoma; phosphoproteomics.
© 2022 The Authors. FEBS Open Bio published by John Wiley & Sons Ltd on behalf of Federation of European Biochemical Societies.
Conflict of interest statement
The authors declare no conflict of interest.
Figures








References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
