

CDKL5 deficiency disorder: clinical features, diagnosis, and management
- PMID: 35483386
- PMCID: PMC9788833
- DOI: 10.1016/S1474-4422(22)00035-7
CDKL5 deficiency disorder: clinical features, diagnosis, and management
Abstract
CDKL5 deficiency disorder (CDD) was first identified as a cause of human disease in 2004. Although initially considered a variant of Rett syndrome, CDD is now recognised as an independent disorder and classified as a developmental epileptic encephalopathy. It is characterised by early-onset (generally within the first 2 months of life) seizures that are usually refractory to polypharmacy. Development is severely impaired in patients with CDD, with only a quarter of girls and a smaller proportion of boys achieving independent walking; however, there is clinical variability, which is probably genetically determined. Gastrointestinal, sleep, and musculoskeletal problems are common in CDD, as in other developmental epileptic encephalopathies, but the prevalence of cerebral visual impairment appears higher in CDD. Clinicians diagnosing infants with CDD need to be familiar with the complexities of this disorder to provide appropriate counselling to the patients' families. Despite some benefit from ketogenic diets and vagal nerve stimulation, there has been little evidence that conventional antiseizure medications or their combinations are helpful in CDD, but further treatment trials are finally underway.
Copyright © 2022 Elsevier Ltd. All rights reserved.
Conflict of interest statement
Declaration of interests HL has received support from the Australian National Health and Medical Research Council (NHMRC; NHMRC Senior Research Fellowship [number 1117105]) in relation to this Review; has received funding from the NIH, the International Foundation for CDKL5 Research, the Orphan Disease Centre, the University of Pennsylvania, and Marinus in relation to this subject matter; and has consulted for Ovid Therapeutics on a related subject matter. JD has received funding from the NIH, the International Foundation for CDKL5 Research, the Orphan Disease Centre, the University of Pennsylvania, and Marinus in relation to this subject matter and has consulted for Ovid Therapeutics on a related subject matter. TAB has received funding from the NIH and the Children's Hospital Foundation for a related subject matter and has consulted for Neuren/Acadia, Ovid/Takeda, AveXis, Marinus Taysha, Alcyone, and Marinus (all compensation was made to his department). SD has funding from the NIH and the International Foundation for CDKL5 Research related to this subject matter; has consulted for Marinus and Ovid Therapeutics on a related subject matter; and is a member of the Scientific Advisory Board for Families SCN2A and SLC6A1 Connect. HO has received funding from the National Institute of Neurological Disorders and Stroke and the International Foundation for CDKL5 Research related to this subject matter; has received funding from the Manton Center for Rare Disease Research and Lou Lou for unrelated research; has consulted for Takeda, Zogenix, and Ovid regarding clinical trials in CDD or information about CDD; and has consulted for the FOXG1 Research Foundation on an unrelated matter to CDD. LS declares no competing interests.
Figures
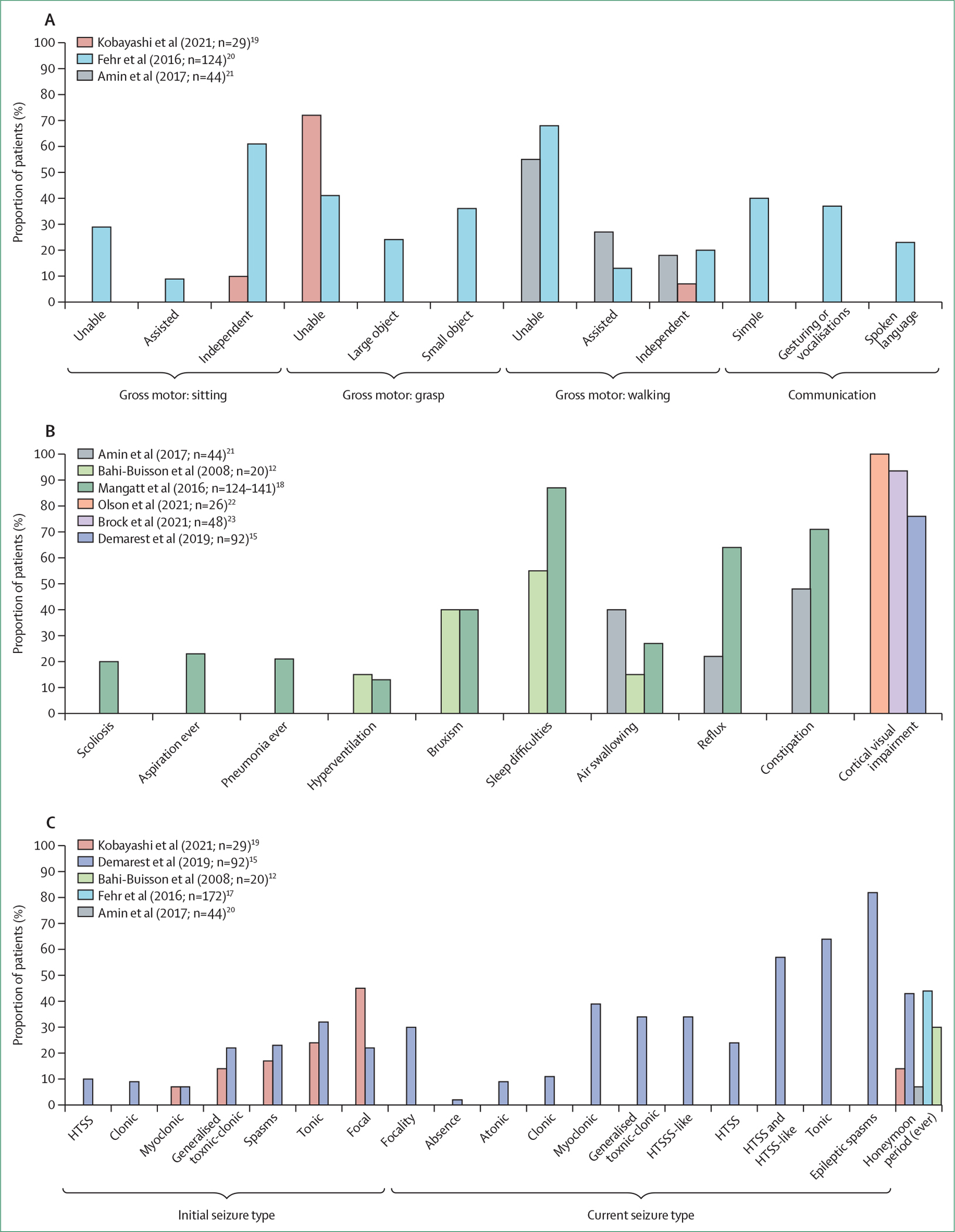
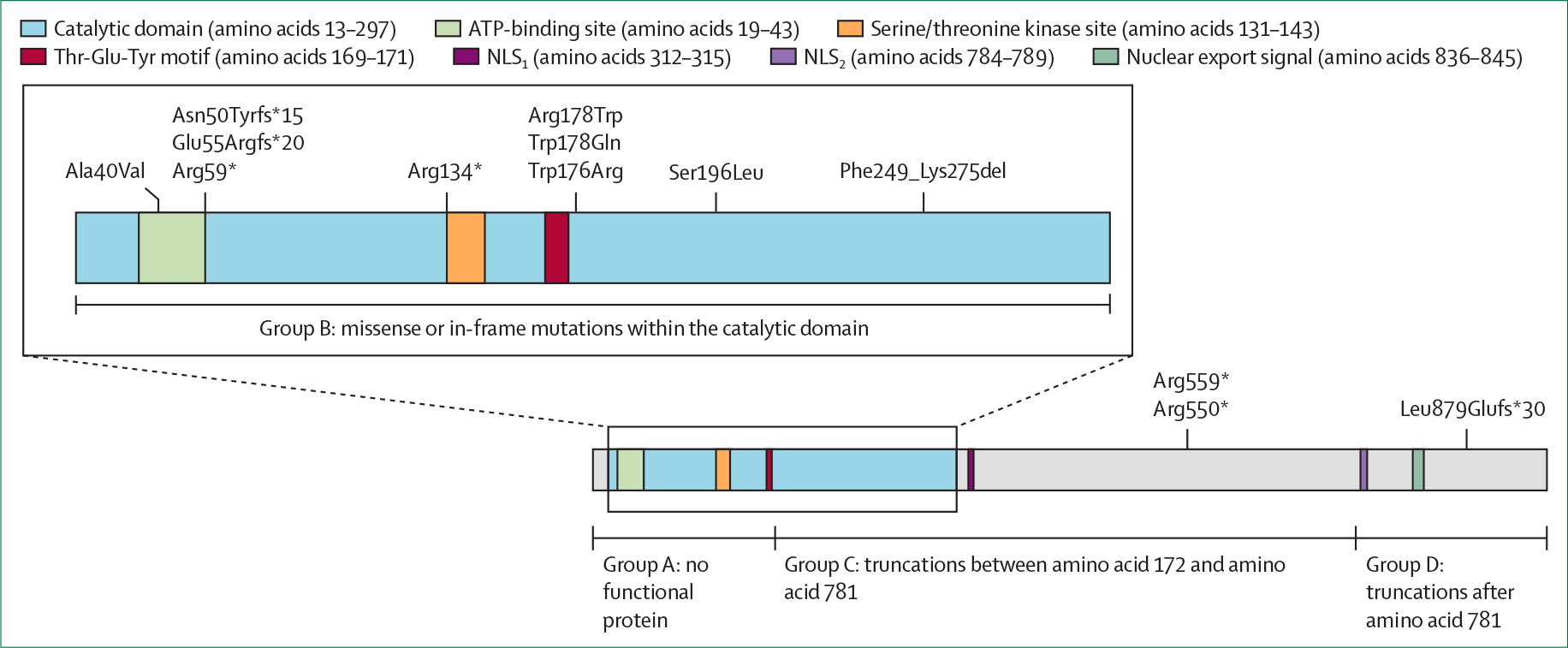
References
-
- Devinsky O, Verducci C, Thiele EA, et al. Open-label use of highly purified CBD (Epidiolex) in patients with CDKL5 deficiency disorder and Aicardi, Dup15q, and Doose syndromes. Epilepsy Behav 2018; 86: 131–37. - PubMed
-
- Hanefeld F The clinical pattern of the Rett syndrome. Brain Dev 1985; 7: 320–25. - PubMed
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
