

Somatic whole genome dynamics of precancer in Barrett's esophagus reveals features associated with disease progression
- PMID: 35484108
- PMCID: PMC9050715
- DOI: 10.1038/s41467-022-29767-7
Somatic whole genome dynamics of precancer in Barrett's esophagus reveals features associated with disease progression
Abstract
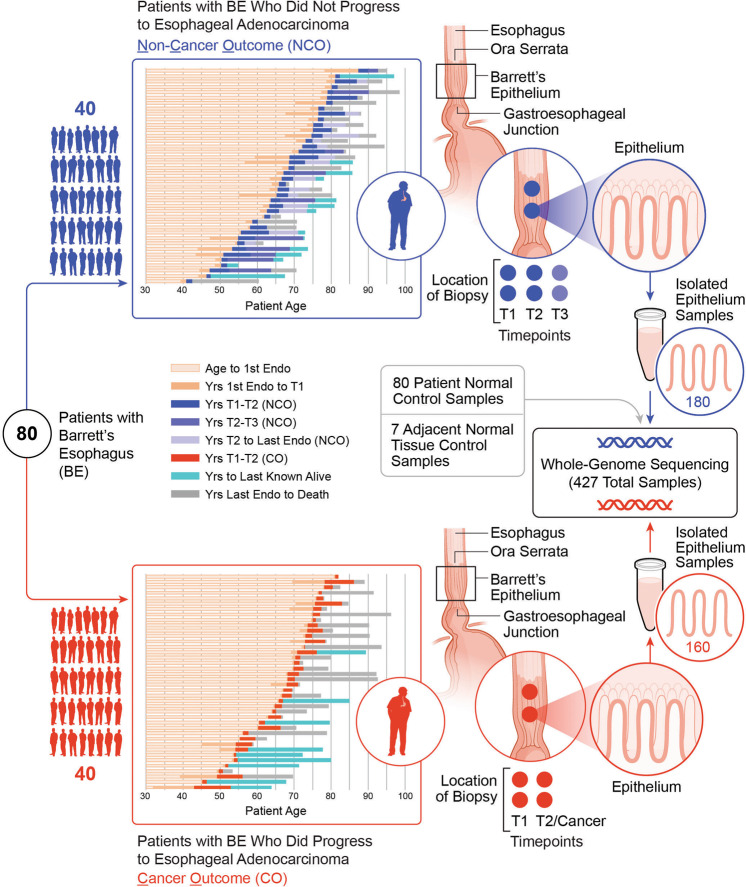
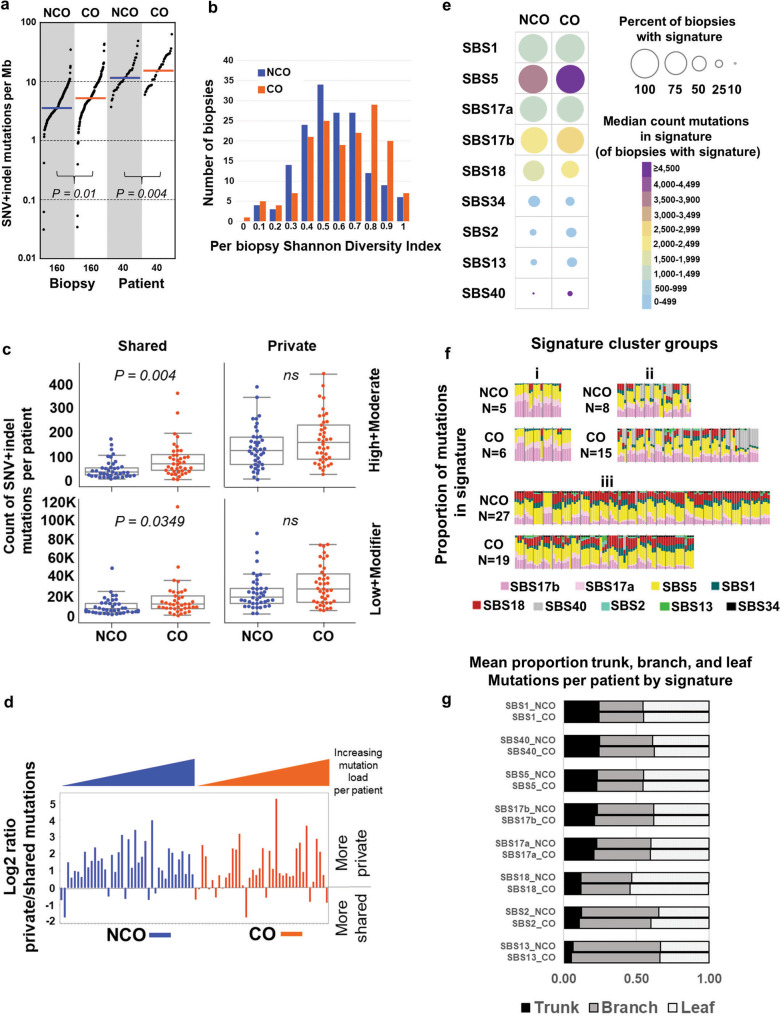
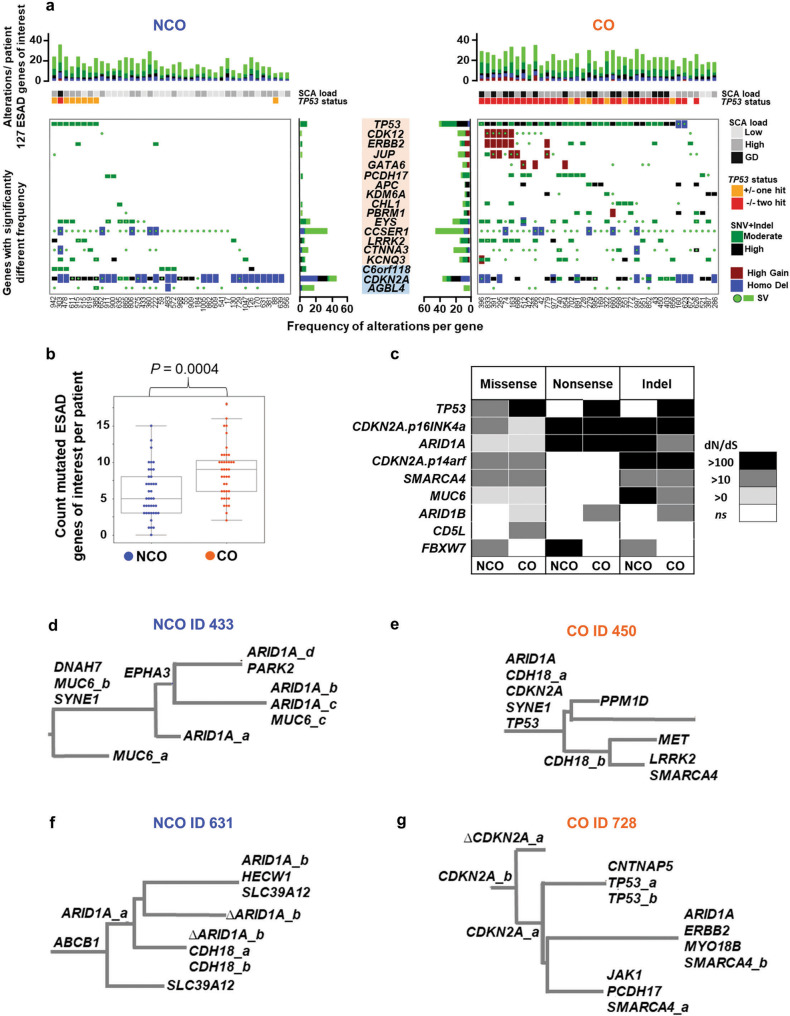
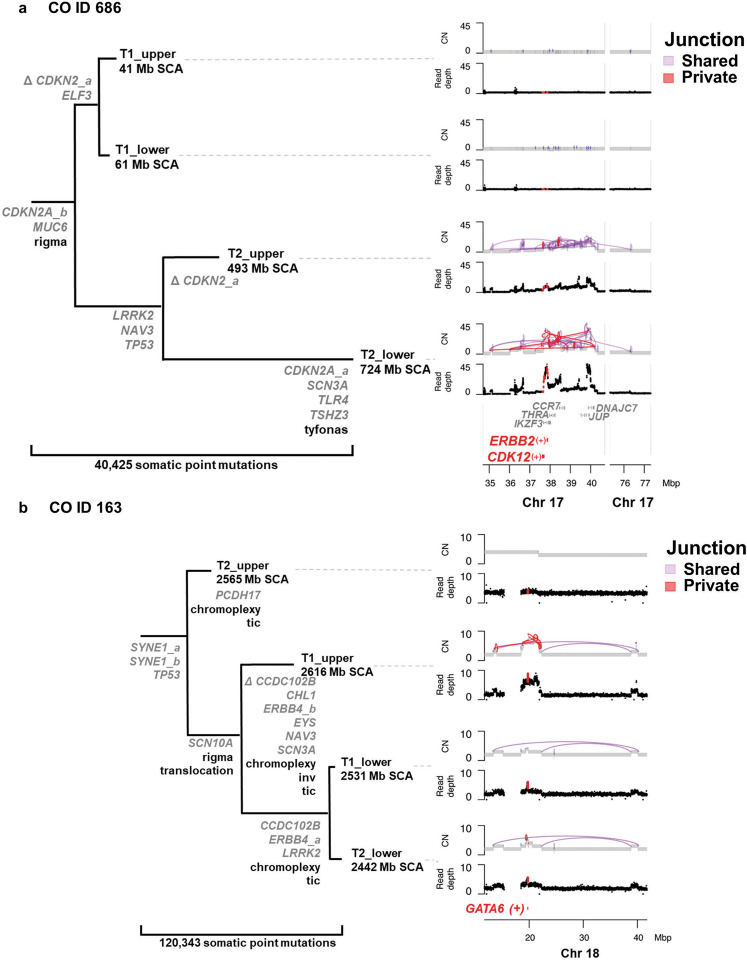
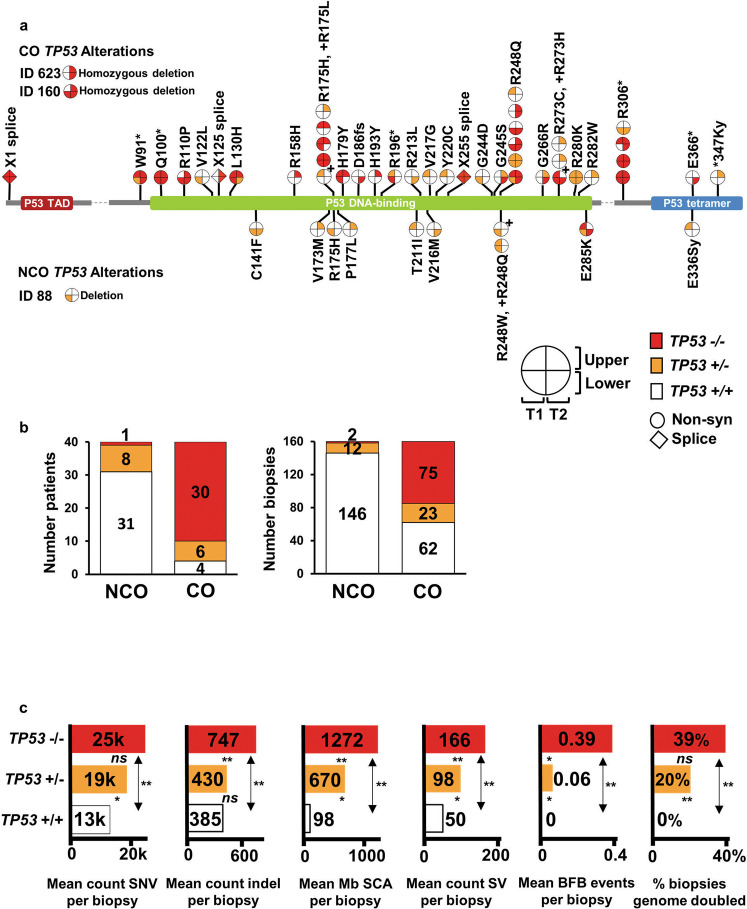
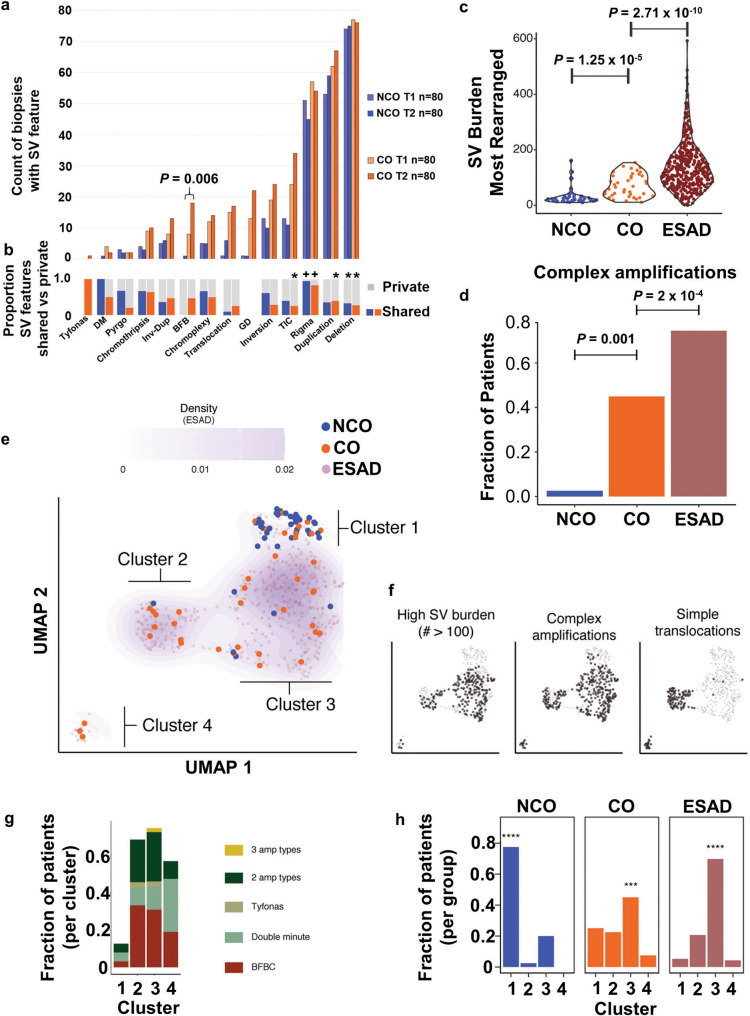
While the genomes of normal tissues undergo dynamic changes over time, little is understood about the temporal-spatial dynamics of genomes in premalignant tissues that progress to cancer compared to those that remain cancer-free. Here we use whole genome sequencing to contrast genomic alterations in 427 longitudinal samples from 40 patients with stable Barrett's esophagus compared to 40 Barrett's patients who progressed to esophageal adenocarcinoma (ESAD). We show the same somatic mutational processes are active in Barrett's tissue regardless of outcome, with high levels of mutation, ESAD gene and focal chromosomal alterations, and similar mutational signatures. The critical distinction between stable Barrett's versus those who progress to cancer is acquisition and expansion of TP53-/- cell populations having complex structural variants and high-level amplifications, which are detectable up to six years prior to a cancer diagnosis. These findings reveal the timing of common somatic genome dynamics in stable Barrett's esophagus and define key genomic features specific to progression to esophageal adenocarcinoma, both of which are critical for cancer prevention and early detection strategies.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
Miscellaneous
