

Deficiency in coatomer complex I causes aberrant activation of STING signalling
- PMID: 35484149
- PMCID: PMC9051092
- DOI: 10.1038/s41467-022-29946-6
Deficiency in coatomer complex I causes aberrant activation of STING signalling
Abstract
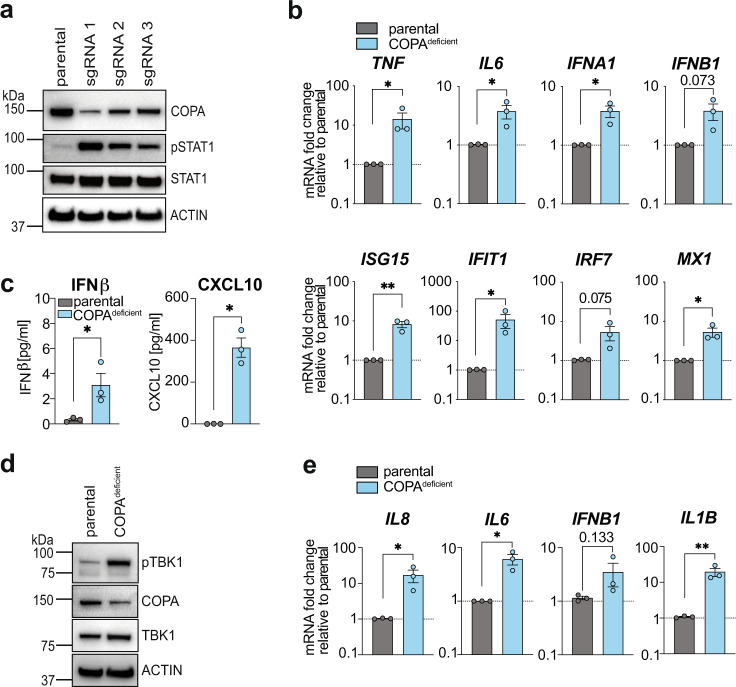
Coatomer complex I (COPI) mediates retrograde vesicular trafficking from Golgi to the endoplasmic reticulum (ER) and within Golgi compartments. Deficiency in subunit alpha causes COPA syndrome and is associated with type I IFN signalling, although the upstream innate immune sensor involved was unknown. Using in vitro models we find aberrant activation of the STING pathway due to deficient retrograde but probably not intra-Golgi transport. Further we find the upstream cytosolic DNA sensor cGAS as essentially required to drive type I IFN signalling. Genetic deletion of COPI subunits COPG1 or COPD similarly induces type I IFN activation in vitro, which suggests that inflammatory diseases associated with mutations in other COPI subunit genes may exist. Finally, we demonstrate that inflammation in COPA syndrome patient peripheral blood mononuclear cells and COPI-deficient cell lines is ameliorated by treatment with the small molecule STING inhibitor H-151, suggesting targeted inhibition of the cGAS/STING pathway as a promising therapeutic approach.
© 2022. The Author(s).
Conflict of interest statement
M.Ga. declares consultancy and speakers fee from Novartis and SOBI. S.L.M. receives funding from IFM therapeutics and M.Ge. consults to IFM Therapeutics. The other authors declare no competing interests.
Figures





References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
