

Fibroblast growth factor receptor 4 promotes glioblastoma progression: a central role of integrin-mediated cell invasiveness
- PMID: 35484633
- PMCID: PMC9052585
- DOI: 10.1186/s40478-022-01363-2
Fibroblast growth factor receptor 4 promotes glioblastoma progression: a central role of integrin-mediated cell invasiveness
Abstract
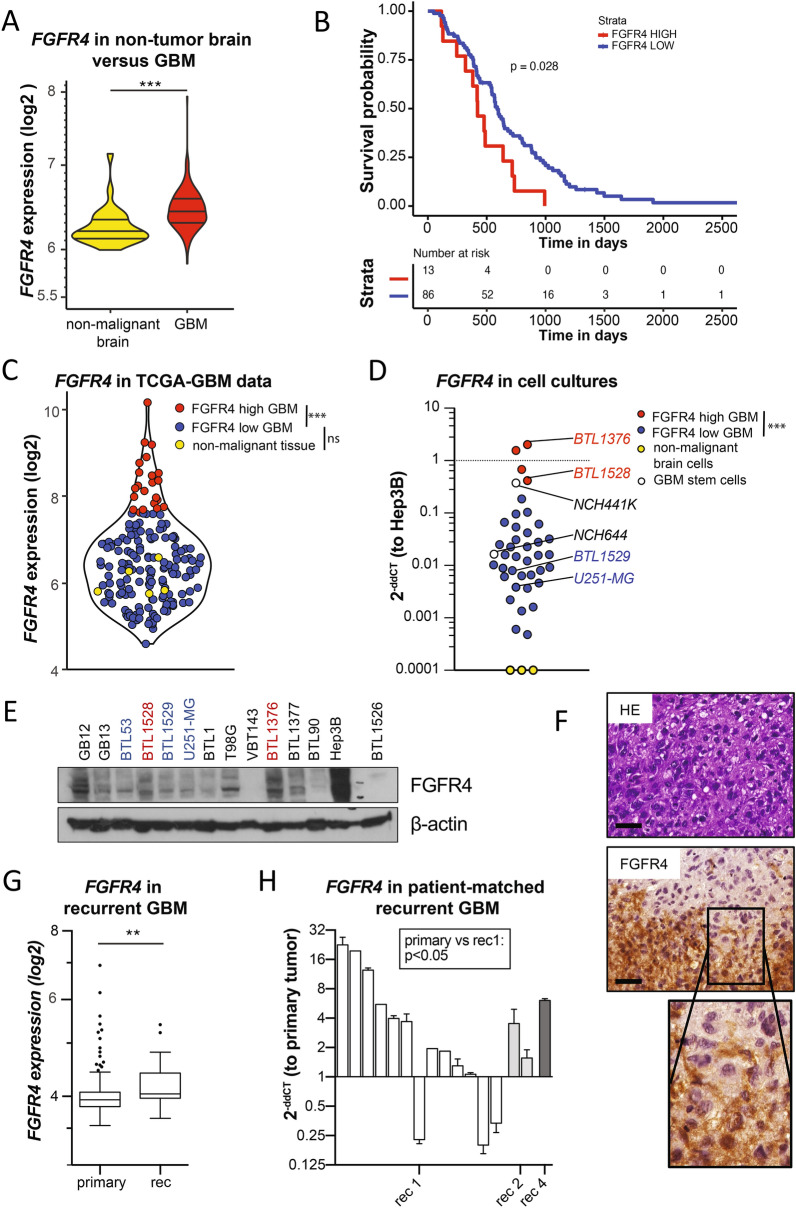
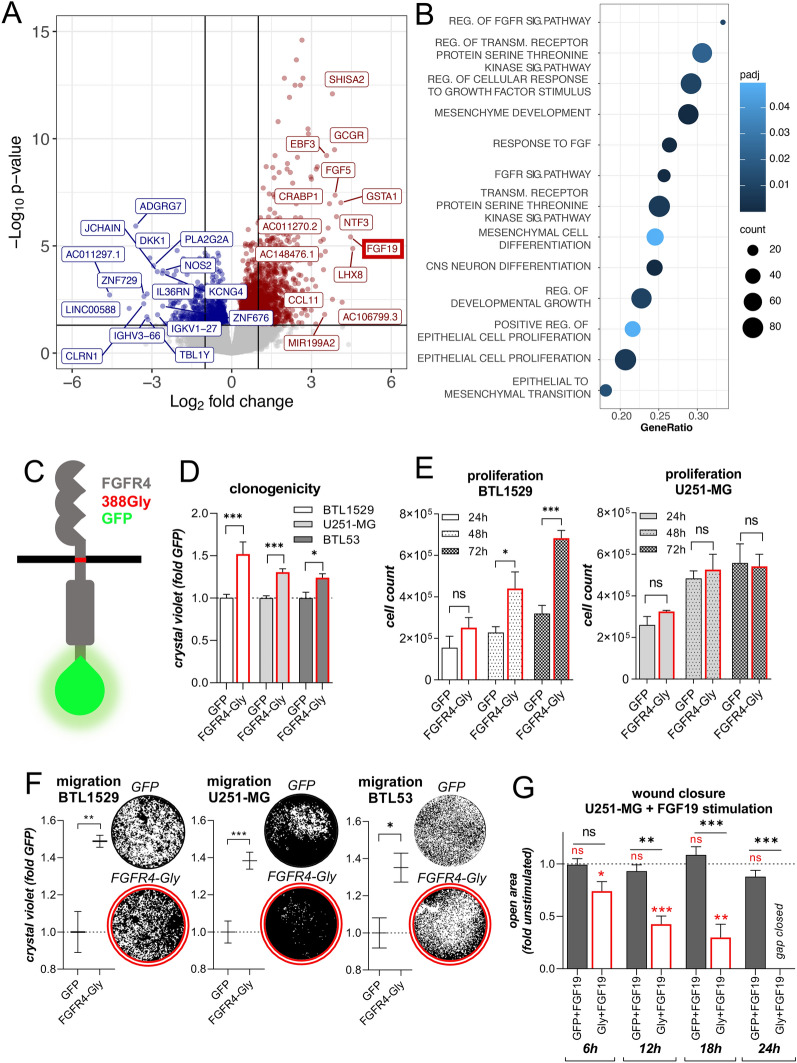
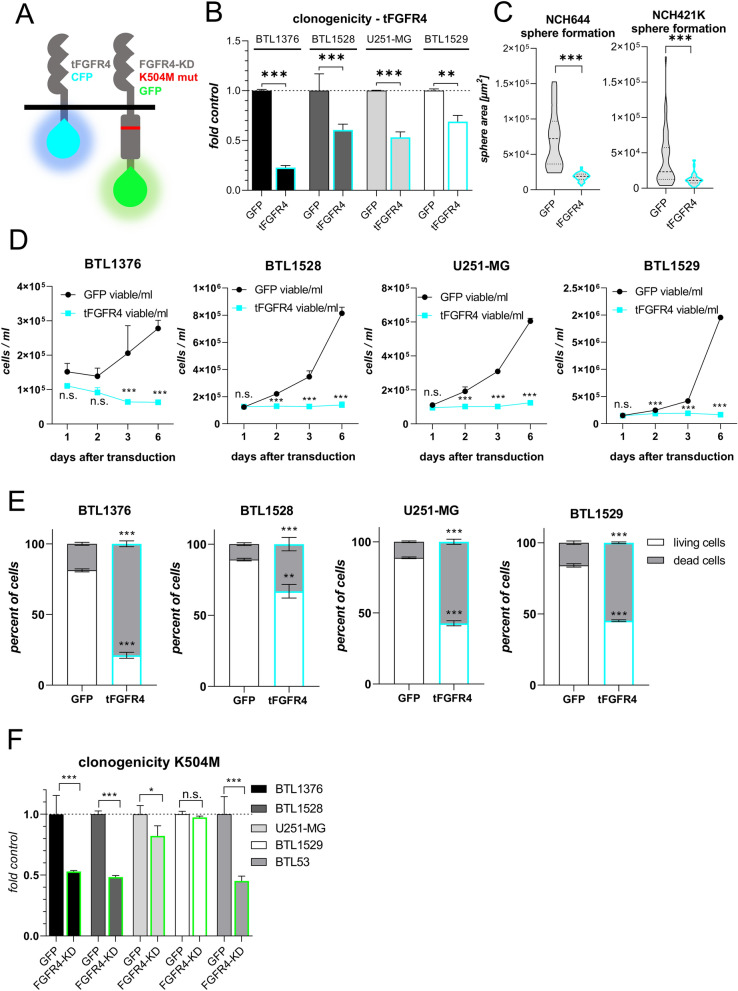
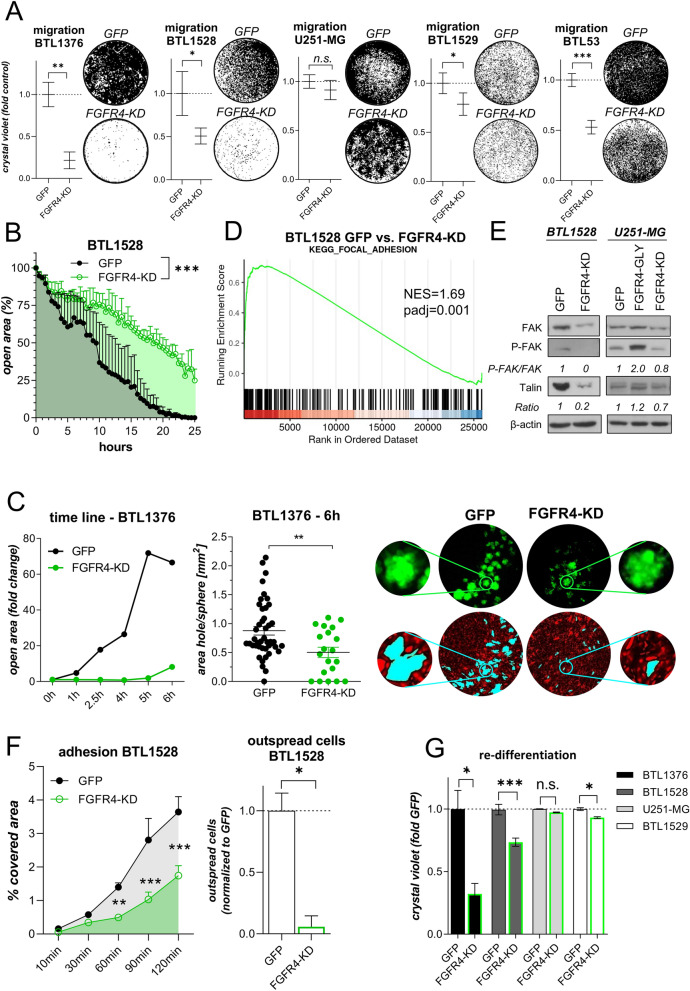
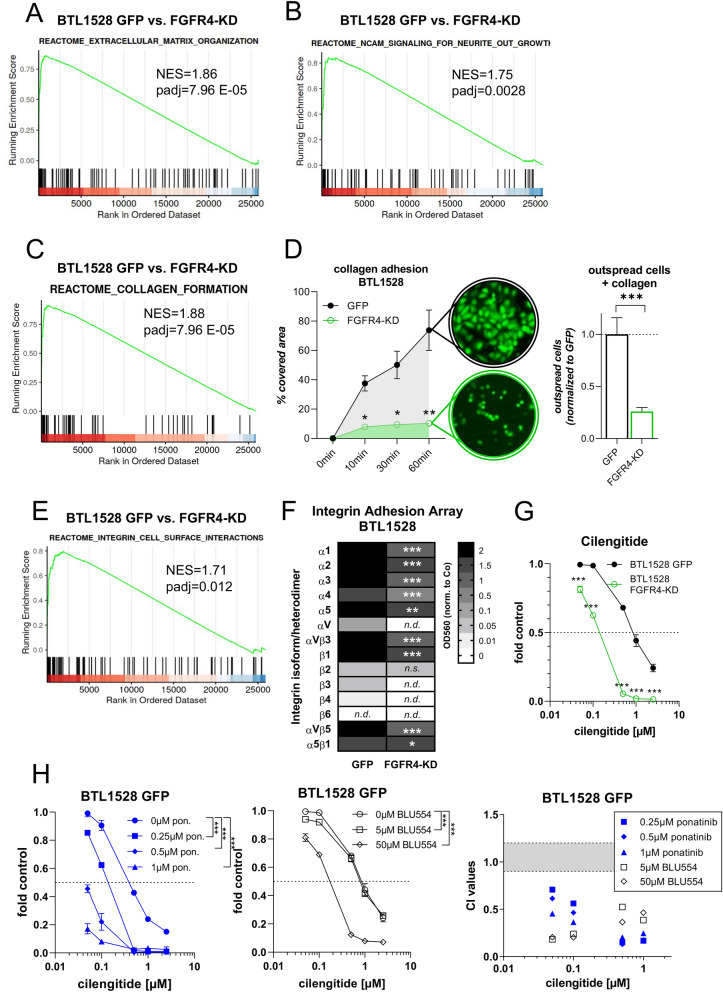
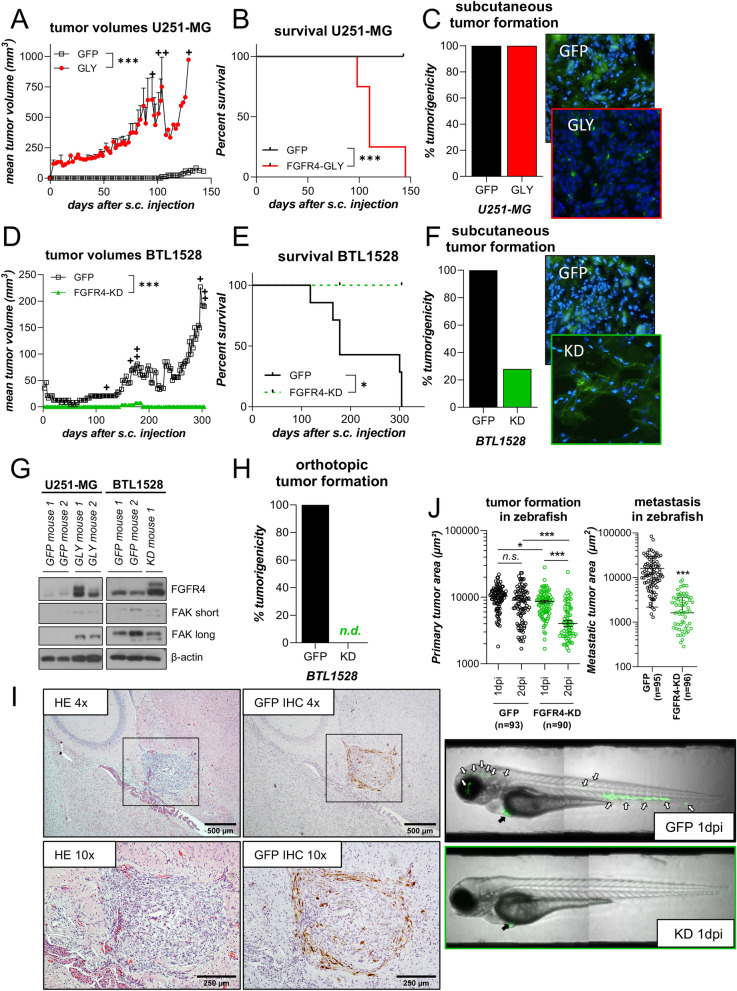
Glioblastoma (GBM) is characterized by a particularly invasive phenotype, supported by oncogenic signals from the fibroblast growth factor (FGF)/ FGF receptor (FGFR) network. However, a possible role of FGFR4 remained elusive so far. Several transcriptomic glioma datasets were analyzed. An extended panel of primary surgical specimen-derived and immortalized GBM (stem)cell models and original tumor tissues were screened for FGFR4 expression. GBM models engineered for wild-type and dominant-negative FGFR4 overexpression were investigated regarding aggressiveness and xenograft formation. Gene set enrichment analyses of FGFR4-modulated GBM models were compared to patient-derived datasets. Despite widely absent in adult brain, FGFR4 mRNA was distinctly expressed in embryonic neural stem cells and significantly upregulated in glioblastoma. Pronounced FGFR4 overexpression defined a distinct GBM patient subgroup with dismal prognosis. Expression levels of FGFR4 and its specific ligands FGF19/FGF23 correlated both in vitro and in vivo and were progressively upregulated in the vast majority of recurrent tumors. Based on overexpression/blockade experiments in respective GBM models, a central pro-oncogenic function of FGFR4 concerning viability, adhesion, migration, and clonogenicity was identified. Expression of dominant-negative FGFR4 resulted in diminished (subcutaneous) or blocked (orthotopic) GBM xenograft formation in the mouse and reduced invasiveness in zebrafish xenotransplantation models. In vitro and in vivo data consistently revealed distinct FGFR4 and integrin/extracellular matrix interactions. Accordingly, FGFR4 blockade profoundly sensitized FGFR4-overexpressing GBM models towards integrin/focal adhesion kinase inhibitors. Collectively, FGFR4 overexpression contributes to the malignant phenotype of a highly aggressive GBM subgroup and is associated with integrin-related therapeutic vulnerabilities.
Keywords: FAK; FGF19; FGFR4; Glioblastoma; Integrins; Invasiveness.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
