

Dynamics and Impacts of Transposable Element Proliferation in the Drosophila nasuta Species Group Radiation
- PMID: 35485457
- PMCID: PMC9075770
- DOI: 10.1093/molbev/msac080
Dynamics and Impacts of Transposable Element Proliferation in the Drosophila nasuta Species Group Radiation
Abstract
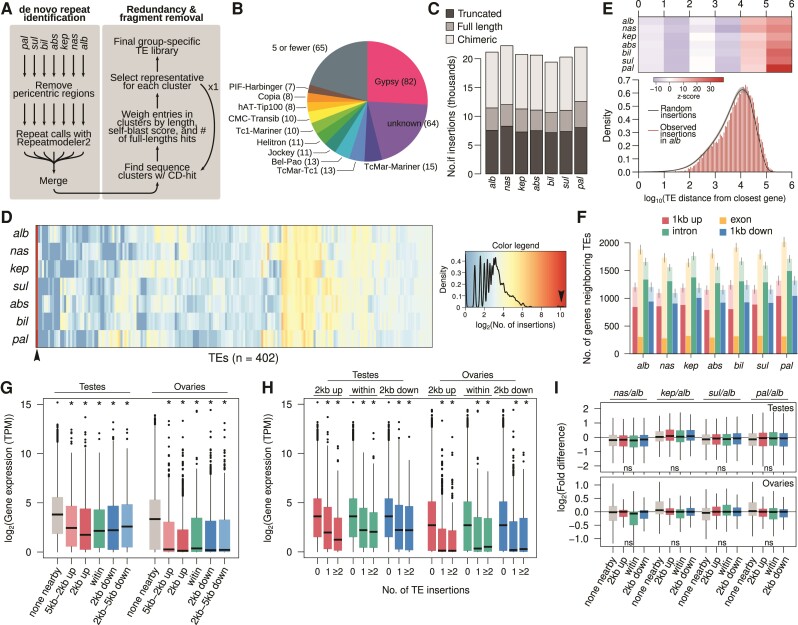
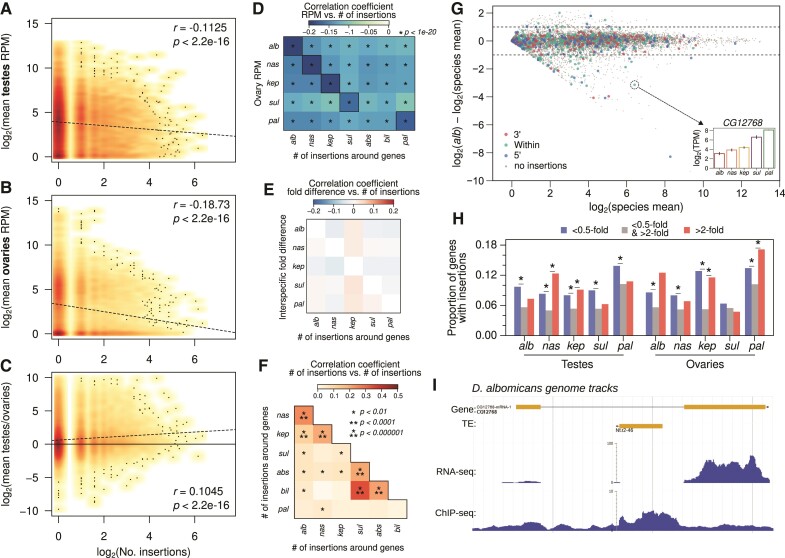
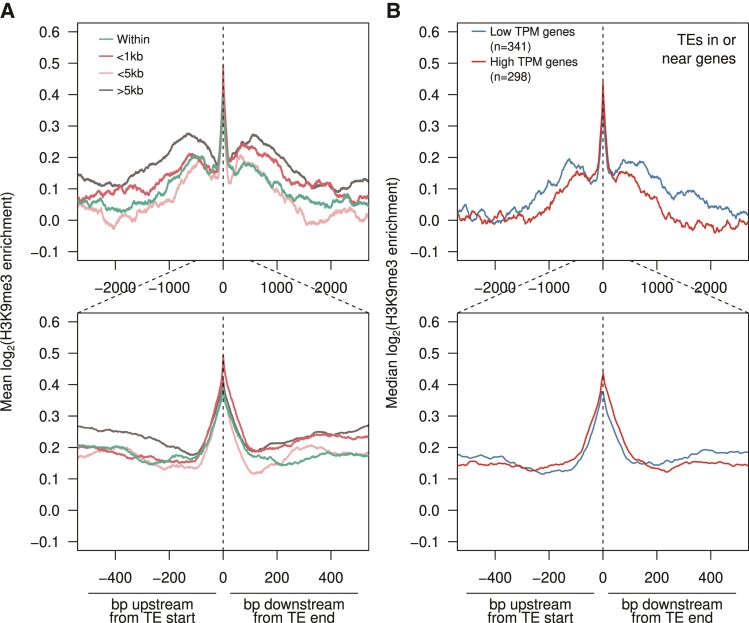
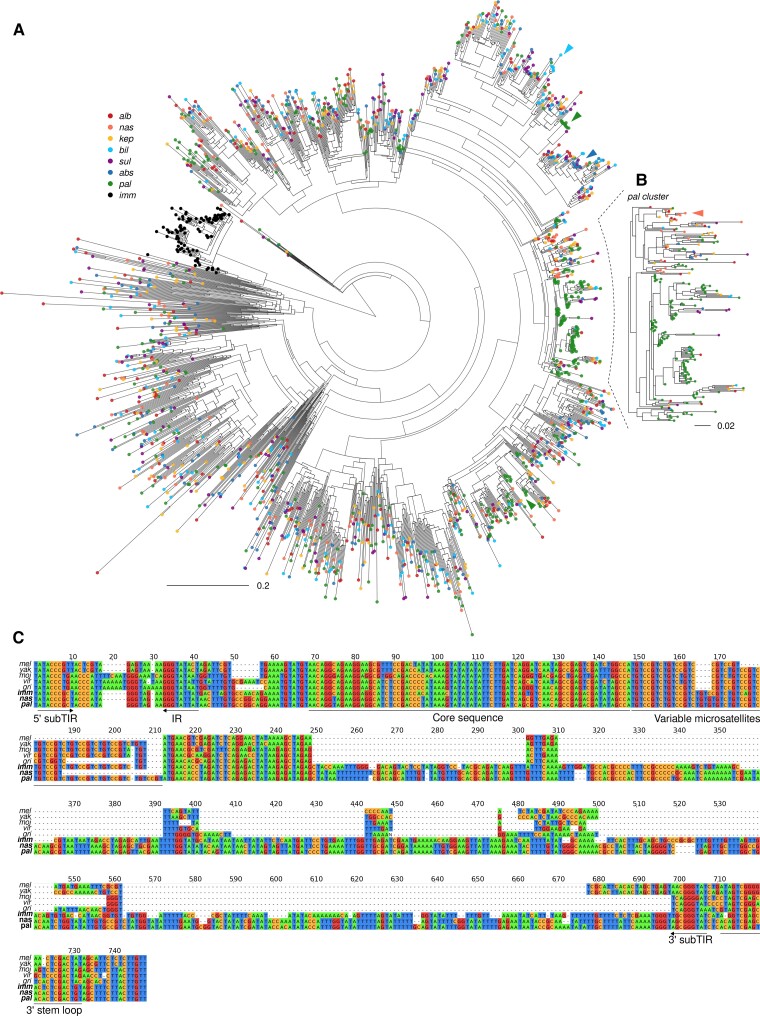
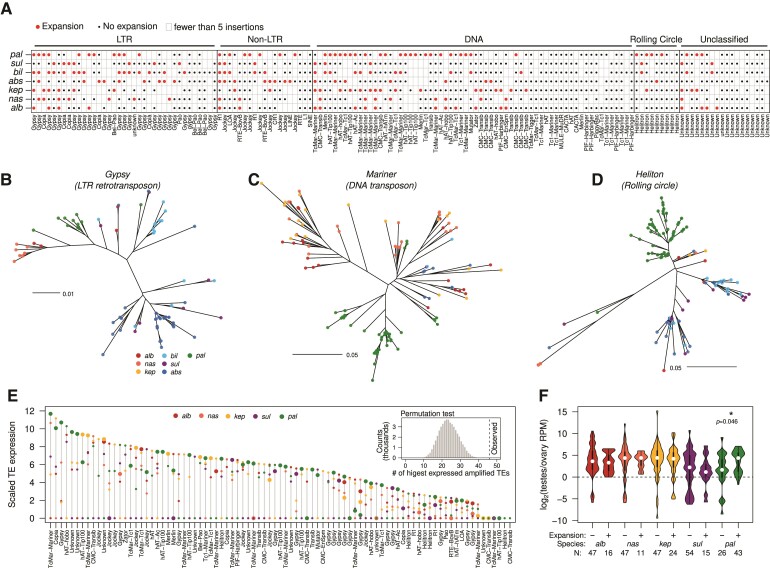
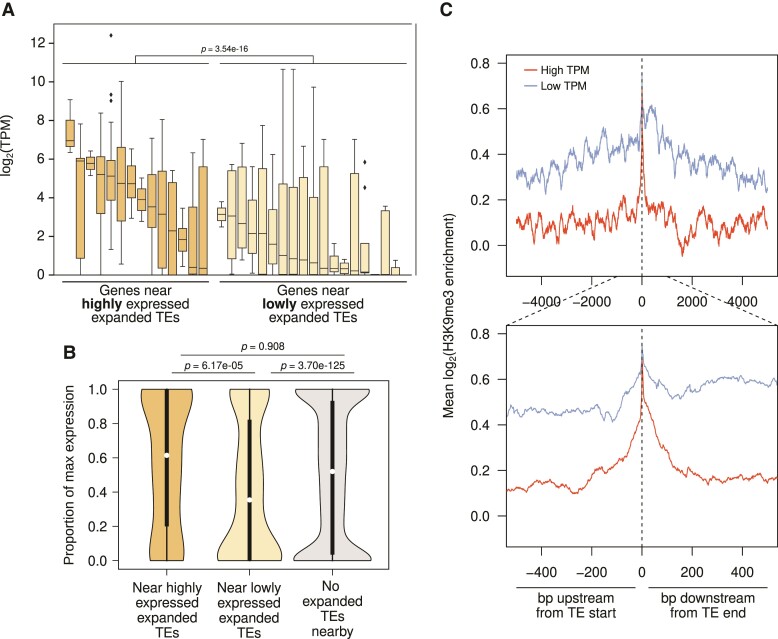
Transposable element (TE) mobilization is a constant threat to genome integrity. Eukaryotic organisms have evolved robust defensive mechanisms to suppress their activity, yet TEs can escape suppression and proliferate, creating strong selective pressure for host defense to adapt. This genomic conflict fuels a never-ending arms race that drives the rapid evolution of TEs and recurrent positive selection of genes involved in host defense; the latter has been shown to contribute to postzygotic hybrid incompatibility. However, how TE proliferation impacts genome and regulatory divergence remains poorly understood. Here, we report the highly complete and contiguous (N50 = 33.8-38.0 Mb) genome assemblies of seven closely related Drosophila species that belong to the nasuta species group-a poorly studied group of flies that radiated in the last 2 My. We constructed a high-quality de novo TE library and gathered germline RNA-seq data, which allowed us to comprehensively annotate and compare TE insertion patterns between the species, and infer the evolutionary forces controlling their spread. We find a strong negative association between TE insertion frequency and expression of genes nearby; this likely reflects survivor bias from reduced fitness impact of TEs inserting near lowly expressed, nonessential genes, with limited TE-induced epigenetic silencing. Phylogenetic analyses of insertions of 147 TE families reveal that 53% of them show recent amplification in at least one species. The most highly amplified TE is a nonautonomous DNA element (Drosophila INterspersed Element; DINE) which has gone through multiple bouts of expansions with thousands of full-length copies littered throughout each genome. Across all TEs, we find that TEs expansions are significantly associated with high expression in the expanded species consistent with suppression escape. Thus, whereas horizontal transfer followed by the invasion of a naïve genome has been highlighted to explain the long-term survival of TEs, our analysis suggests that evasion of host suppression of resident TEs is a major strategy to persist over evolutionary times. Altogether, our results shed light on the heterogenous and context-dependent nature in which TEs affect gene regulation and the dynamics of rampant TE proliferation amidst a recently radiated species group.
Keywords: Drosophila; epigenetic suppression; transposable elements.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution. All rights reserved. For permissions, please e-mail: journals.permissions@oup.com.
Figures

Similar articles
-
Species-specific chromatin landscape determines how transposable elements shape genome evolution.Elife. 2022 Aug 23;11:e81567. doi: 10.7554/eLife.81567. Elife. 2022. PMID: 35997258 Free PMC article.
-
Abundant and species-specific DINE-1 transposable elements in 12 Drosophila genomes.Genome Biol. 2008;9(2):R39. doi: 10.1186/gb-2008-9-2-r39. Epub 2008 Feb 21. Genome Biol. 2008. PMID: 18291035 Free PMC article.
-
Molecular evolution under increasing transposable element burden in Drosophila: a speed limit on the evolutionary arms race.BMC Evol Biol. 2011 Sep 14;11:258. doi: 10.1186/1471-2148-11-258. BMC Evol Biol. 2011. PMID: 21917173 Free PMC article.
-
Silencing of Transposable Elements by piRNAs in Drosophila: An Evolutionary Perspective.Genomics Proteomics Bioinformatics. 2017 Jun;15(3):164-176. doi: 10.1016/j.gpb.2017.01.006. Epub 2017 Jun 8. Genomics Proteomics Bioinformatics. 2017. PMID: 28602845 Free PMC article. Review.
-
Mammalian transposable elements and their impacts on genome evolution.Chromosome Res. 2018 Mar;26(1-2):25-43. doi: 10.1007/s10577-017-9570-z. Epub 2018 Feb 1. Chromosome Res. 2018. PMID: 29392473 Free PMC article. Review.
Cited by
-
Transposable element and host silencing activity in gigantic genomes.Front Cell Dev Biol. 2023 Feb 24;11:1124374. doi: 10.3389/fcell.2023.1124374. eCollection 2023. Front Cell Dev Biol. 2023. PMID: 36910142 Free PMC article.
-
Evolutionary dynamics between transposable elements and their host genomes: mechanisms of suppression and escape.Curr Opin Genet Dev. 2023 Oct;82:102092. doi: 10.1016/j.gde.2023.102092. Epub 2023 Jul 28. Curr Opin Genet Dev. 2023. PMID: 37517354 Free PMC article. Review.
-
Widespread impact of transposable elements on the evolution of post-transcriptional regulation in the cotton genus Gossypium.Genome Biol. 2025 Mar 17;26(1):60. doi: 10.1186/s13059-025-03534-5. Genome Biol. 2025. PMID: 40098207 Free PMC article.
-
Multiple and diversified transposon lineages contribute to early and recent bivalve genome evolution.BMC Biol. 2023 Jun 26;21(1):145. doi: 10.1186/s12915-023-01632-z. BMC Biol. 2023. PMID: 37365567 Free PMC article.
-
Analyses of 600+ insect genomes reveal repetitive element dynamics and highlight biodiversity-scale repeat annotation challenges.Genome Res. 2023 Oct;33(10):1708-1717. doi: 10.1101/gr.277387.122. Epub 2023 Sep 22. Genome Res. 2023. PMID: 37739812 Free PMC article.
References
-
- Drosophila 12 Genomes Consortium . 2007. Evolution of genes and genomes on the Drosophila phylogeny. Nature 450:203–218. - PubMed
-
- Altschul SF, Gish W, Miller W, Myers EW, Lipman DJ. 1990. Basic local alignment search tool. J Mol Biol. 215:403–410. - PubMed
-
- Anxolabéhère D, Kidwell MG, Periquet G. 1988. Molecular characteristics of diverse populations are consistent with the hypothesis of a recent invasion of Drosophila melanogaster by mobile P elements. Mol Biol Evol. 5:252–269. - PubMed
Publication types
MeSH terms
Substances
Associated data
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Miscellaneous
