

Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline
- PMID: 35486072
- PMCID: PMC9851481
- DOI: 10.1164/rccm.202202-0399ST
Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults: An Official ATS/ERS/JRS/ALAT Clinical Practice Guideline
Abstract
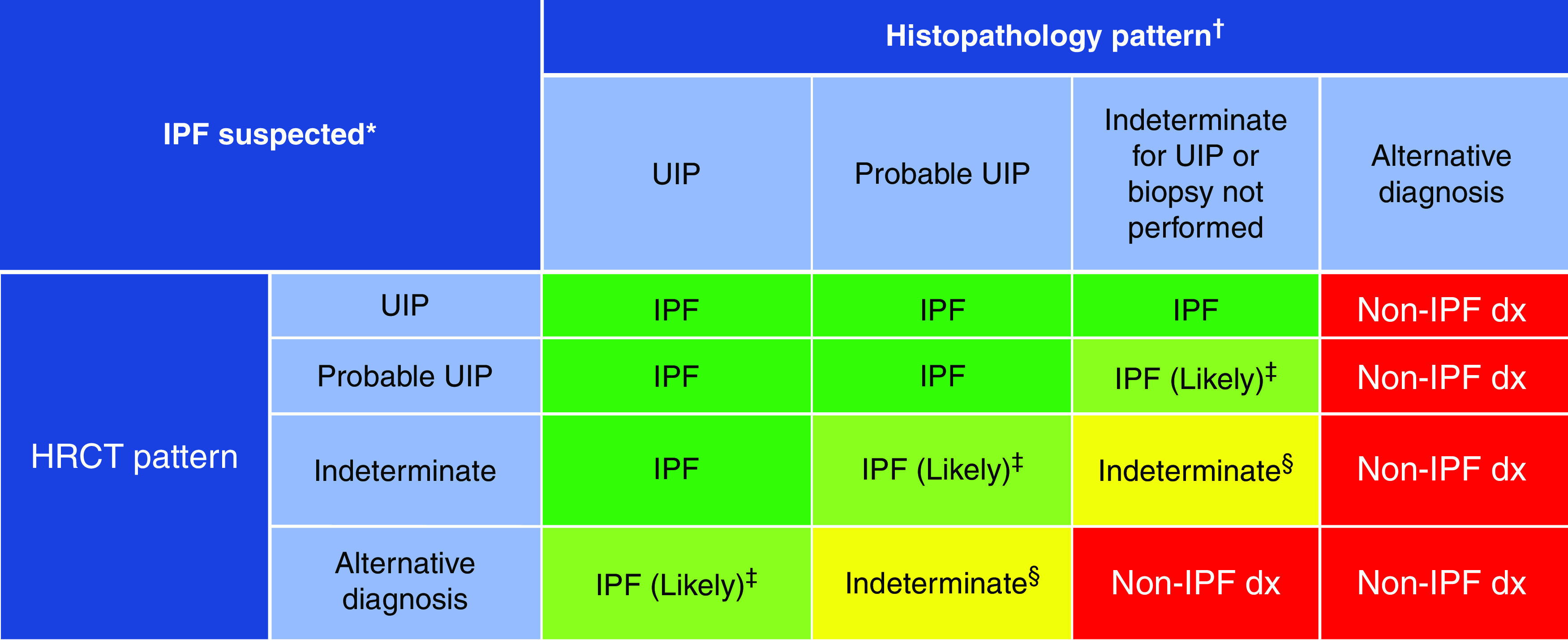
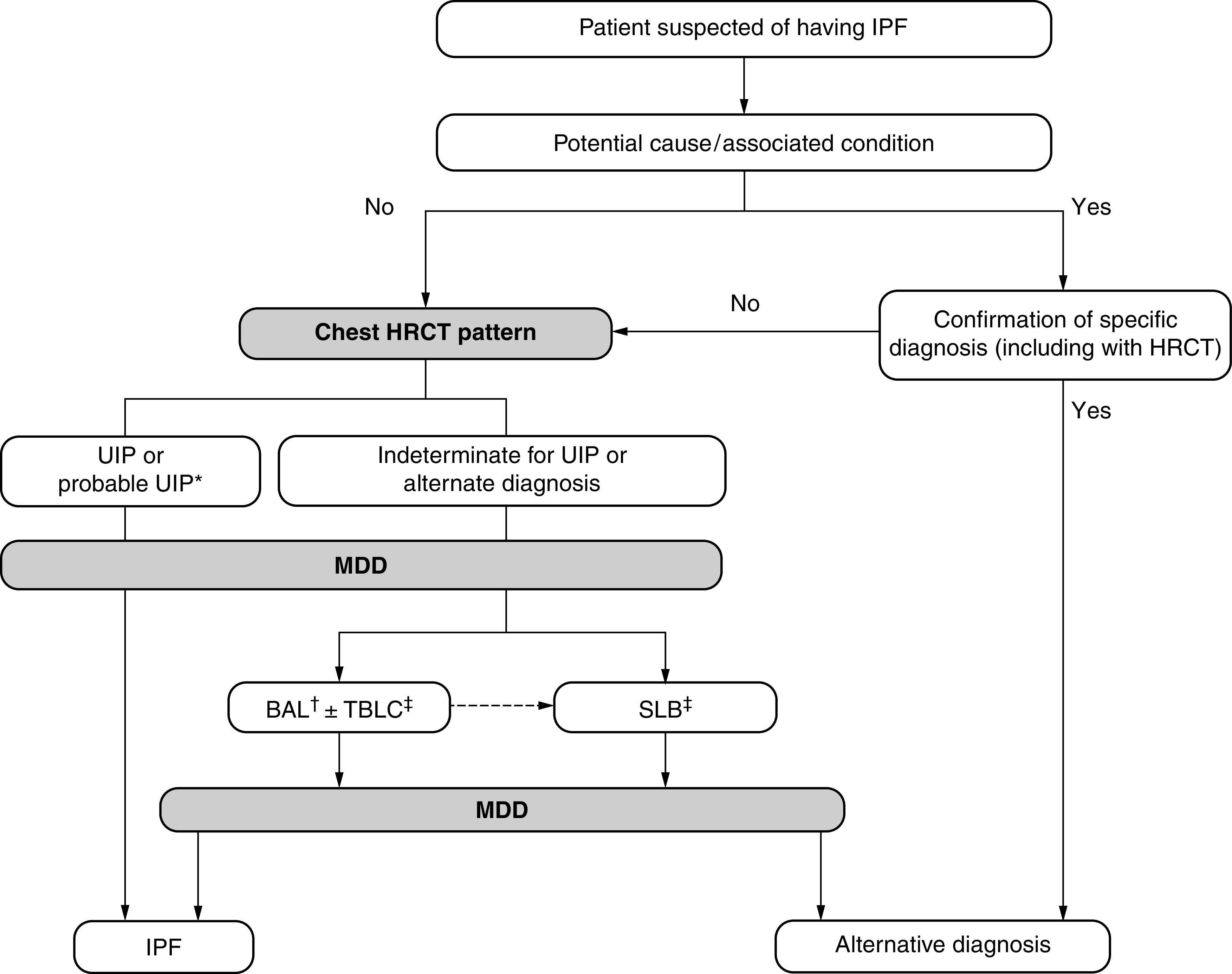
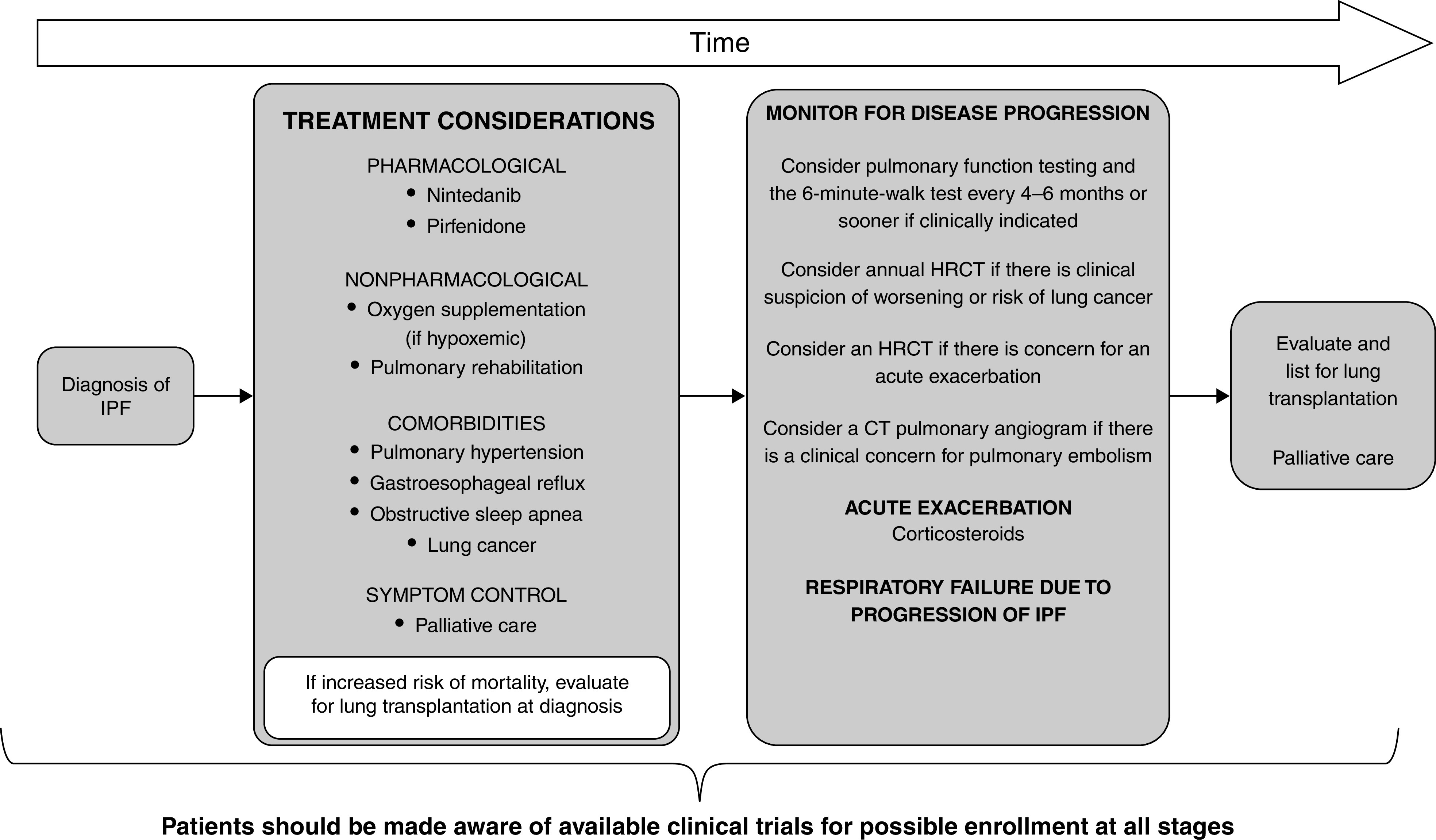
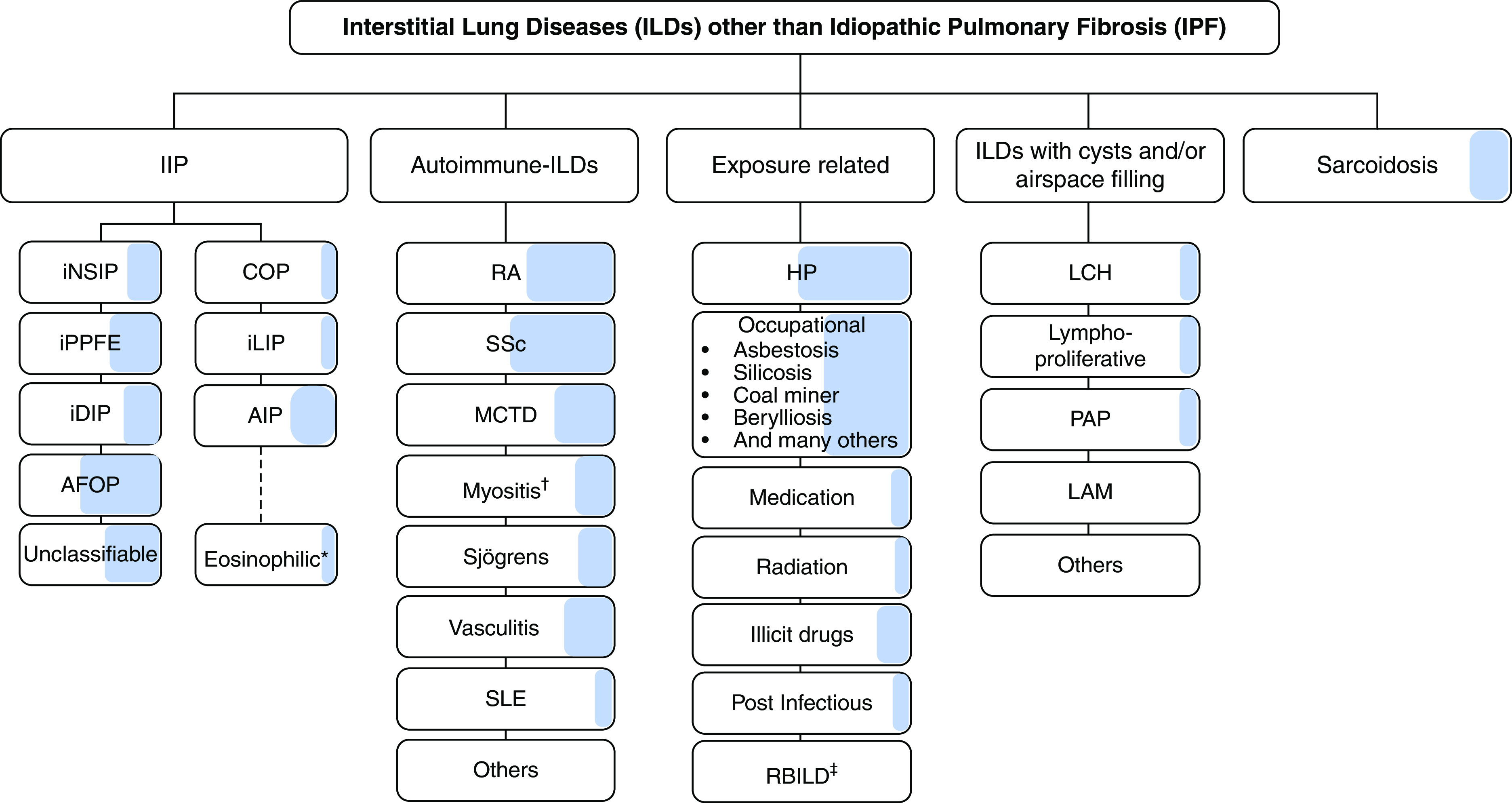
Background: This American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Asociación Latinoamericana de Tórax guideline updates prior idiopathic pulmonary fibrosis (IPF) guidelines and addresses the progression of pulmonary fibrosis in patients with interstitial lung diseases (ILDs) other than IPF. Methods: A committee was composed of multidisciplinary experts in ILD, methodologists, and patient representatives. 1) Update of IPF: Radiological and histopathological criteria for IPF were updated by consensus. Questions about transbronchial lung cryobiopsy, genomic classifier testing, antacid medication, and antireflux surgery were informed by systematic reviews and answered with evidence-based recommendations using the Grading of Recommendations, Assessment, Development and Evaluation (GRADE) approach. 2) Progressive pulmonary fibrosis (PPF): PPF was defined, and then radiological and physiological criteria for PPF were determined by consensus. Questions about pirfenidone and nintedanib were informed by systematic reviews and answered with evidence-based recommendations using the GRADE approach. Results:1) Update of IPF: A conditional recommendation was made to regard transbronchial lung cryobiopsy as an acceptable alternative to surgical lung biopsy in centers with appropriate expertise. No recommendation was made for or against genomic classifier testing. Conditional recommendations were made against antacid medication and antireflux surgery for the treatment of IPF. 2) PPF: PPF was defined as at least two of three criteria (worsening symptoms, radiological progression, and physiological progression) occurring within the past year with no alternative explanation in a patient with an ILD other than IPF. A conditional recommendation was made for nintedanib, and additional research into pirfenidone was recommended. Conclusions: The conditional recommendations in this guideline are intended to provide the basis for rational, informed decisions by clinicians.
Keywords: histopathology; idiopathic pulmonary fibrosis; progressive pulmonary fibrosis; radiology.
Figures
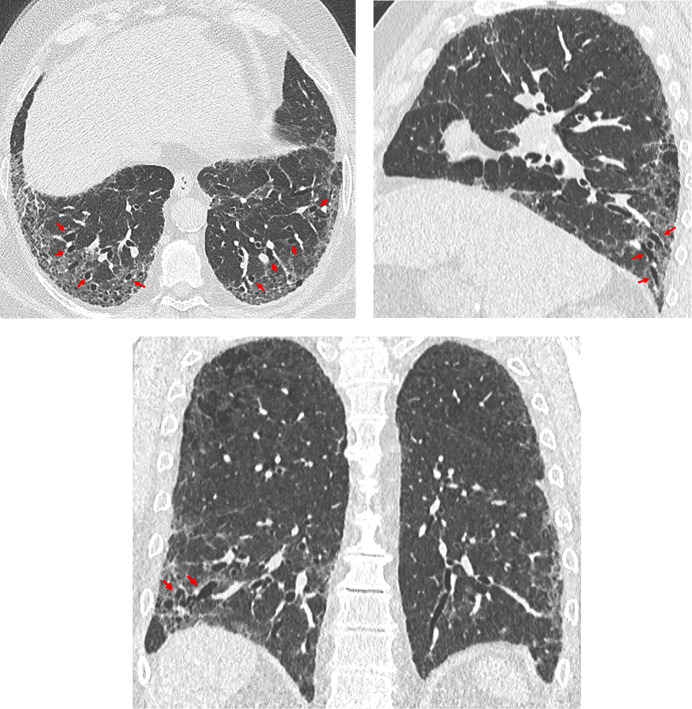
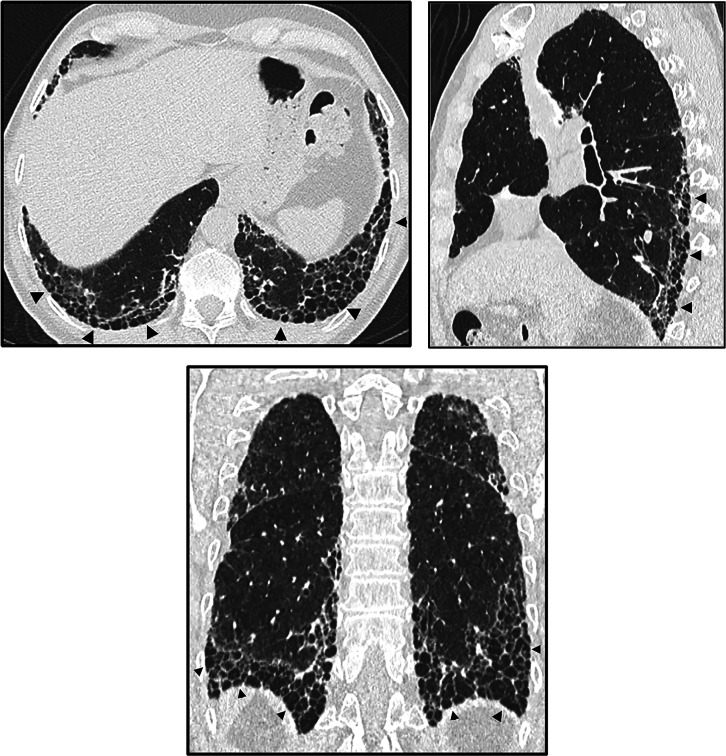
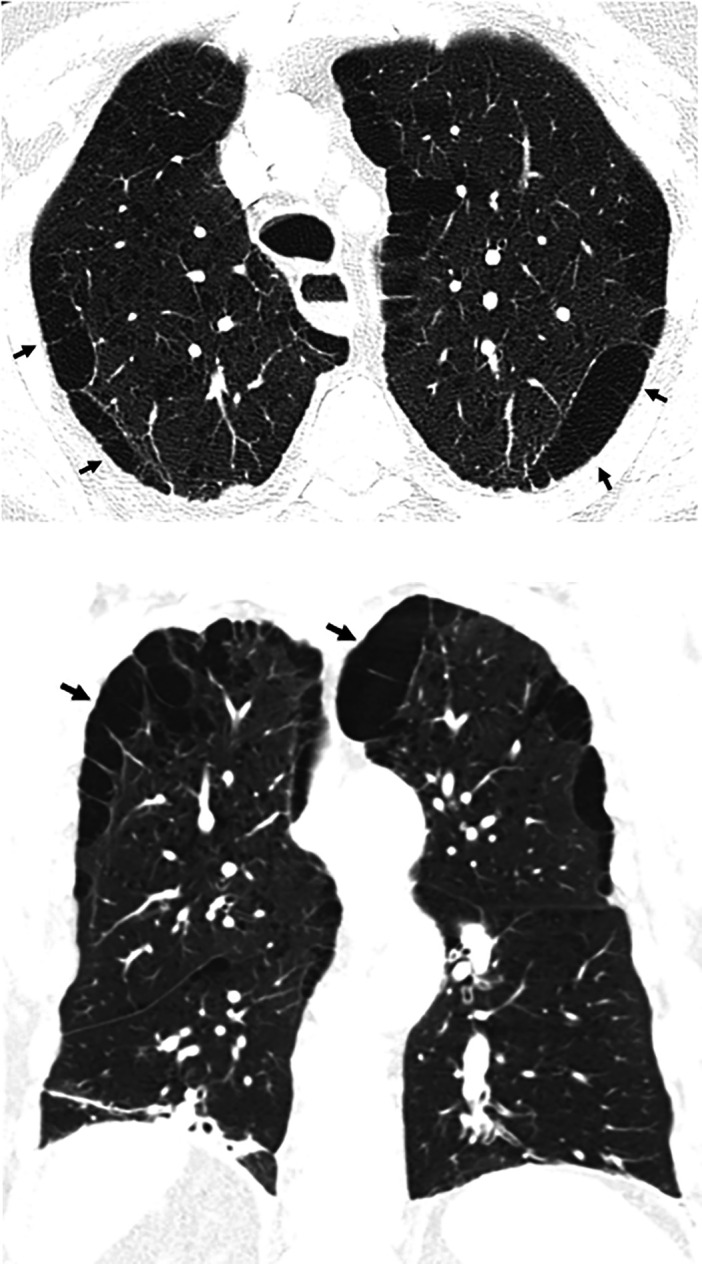
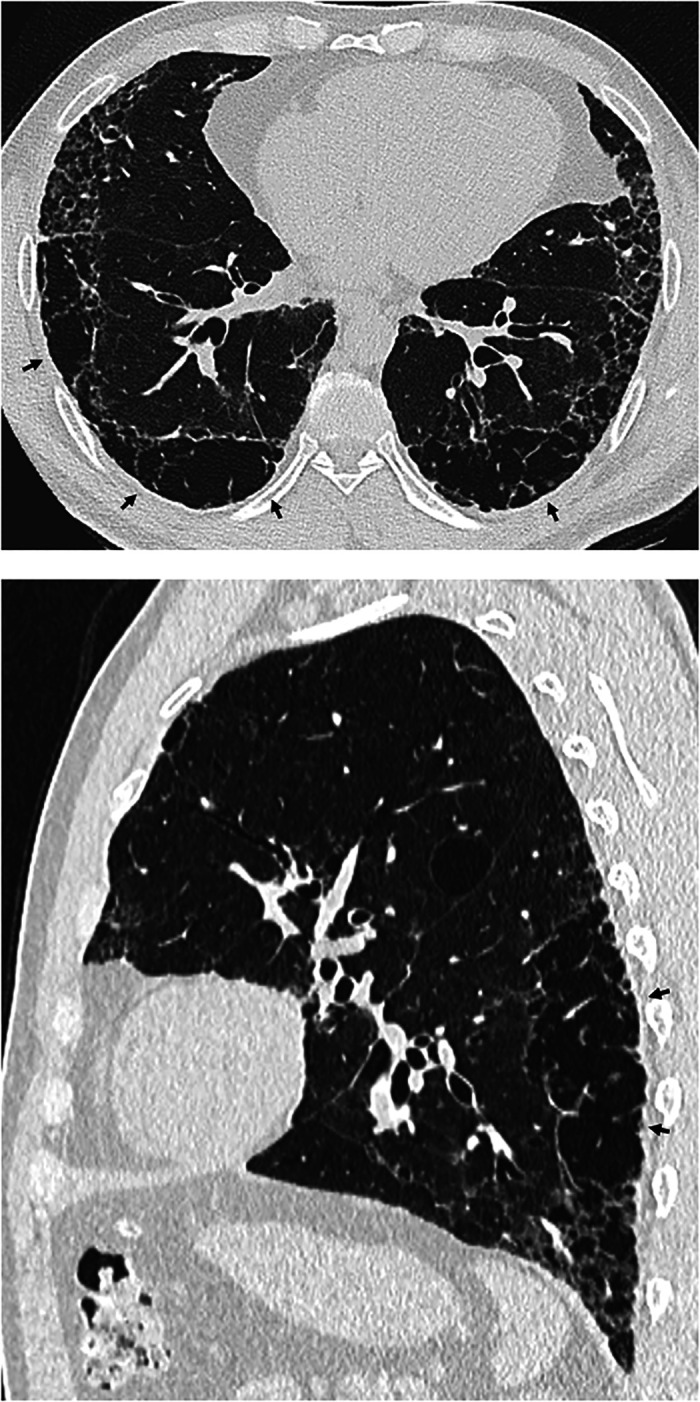
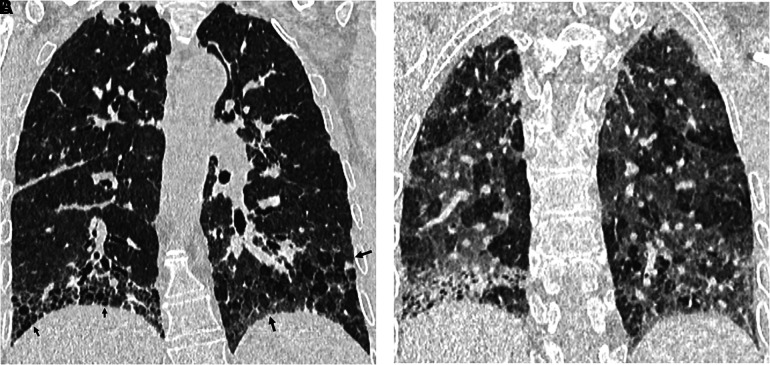
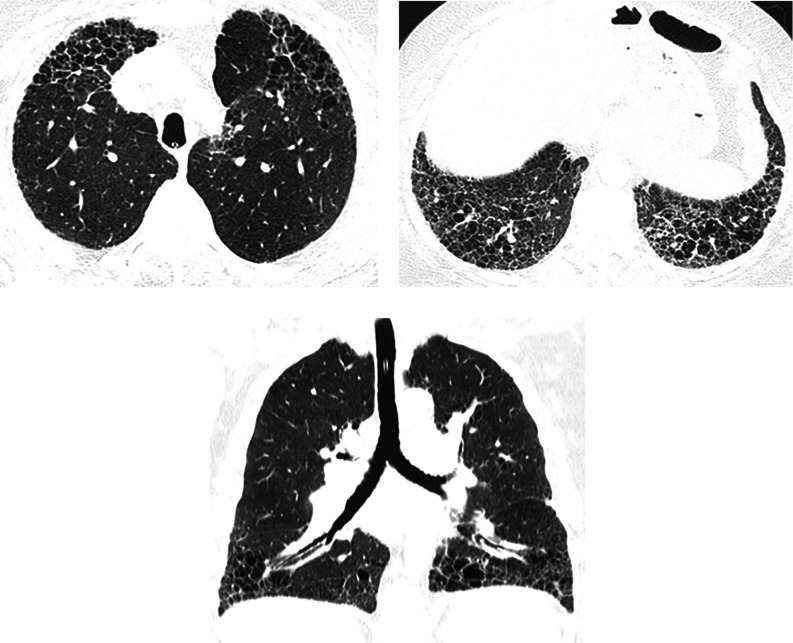
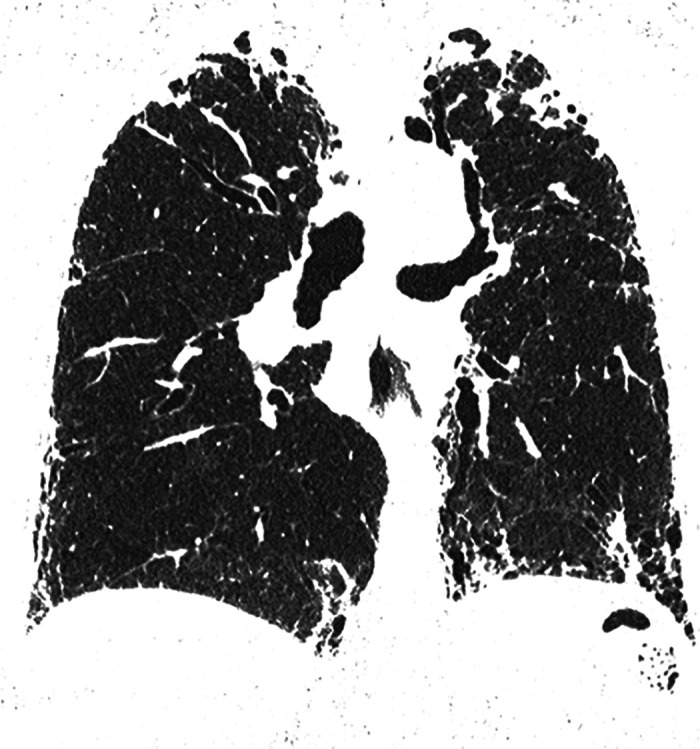
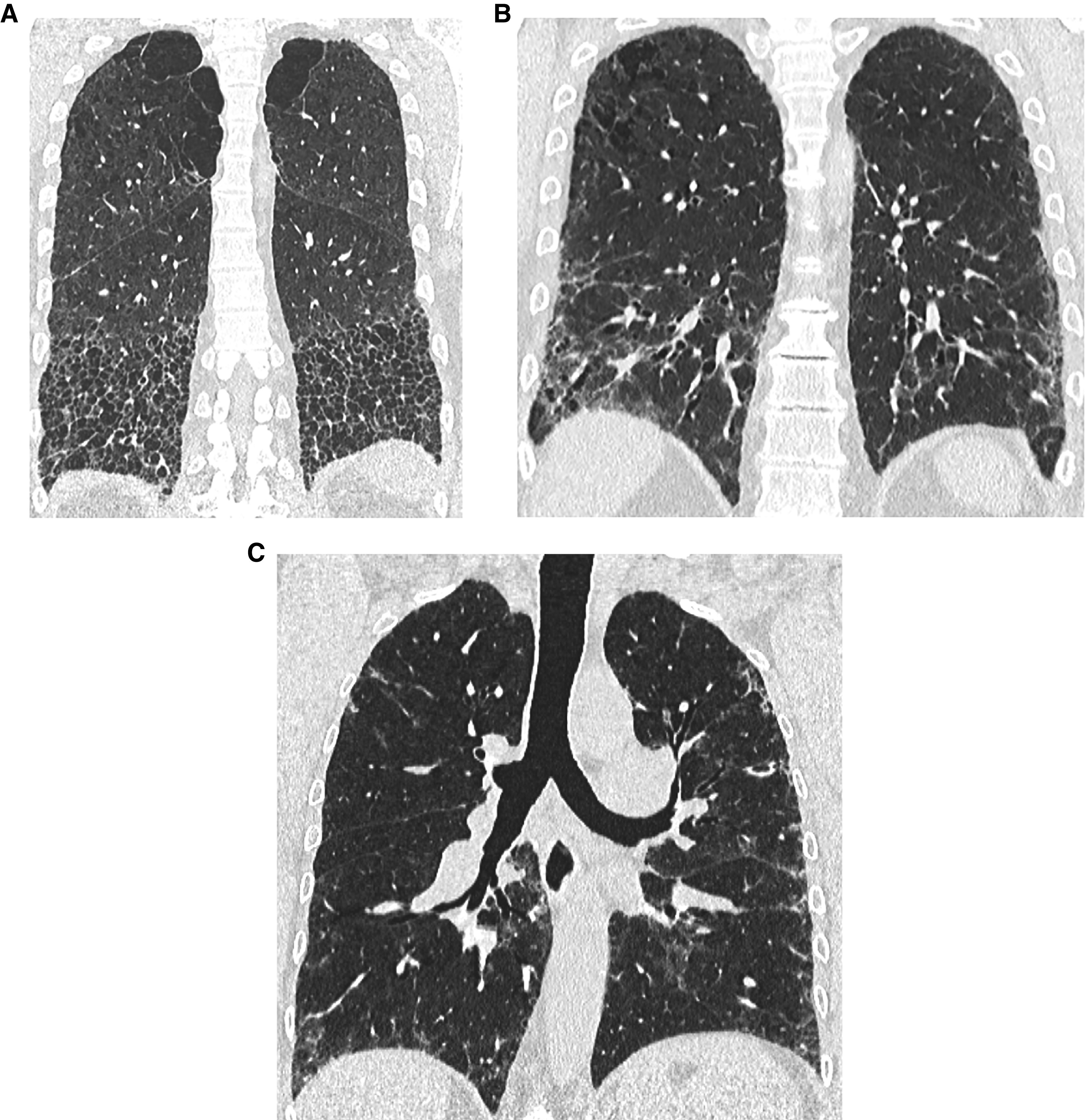
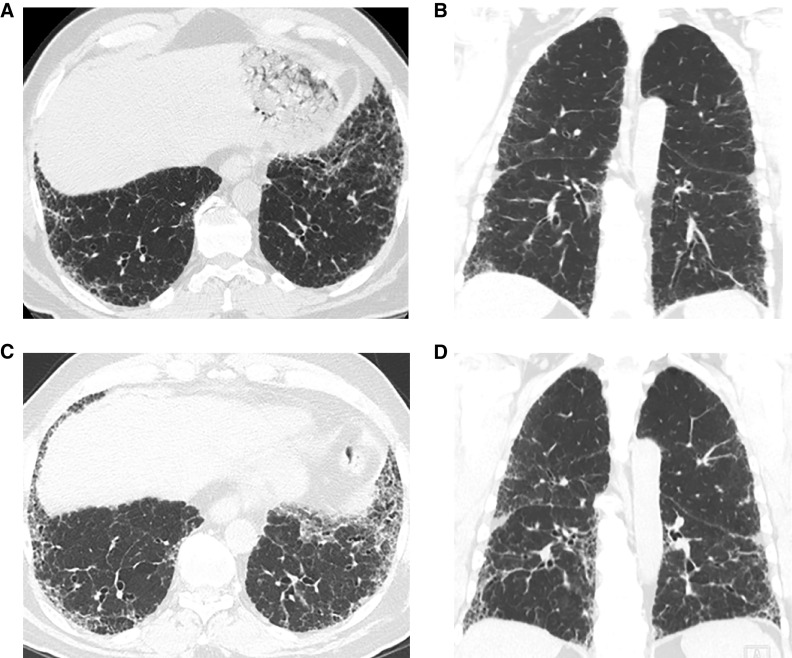
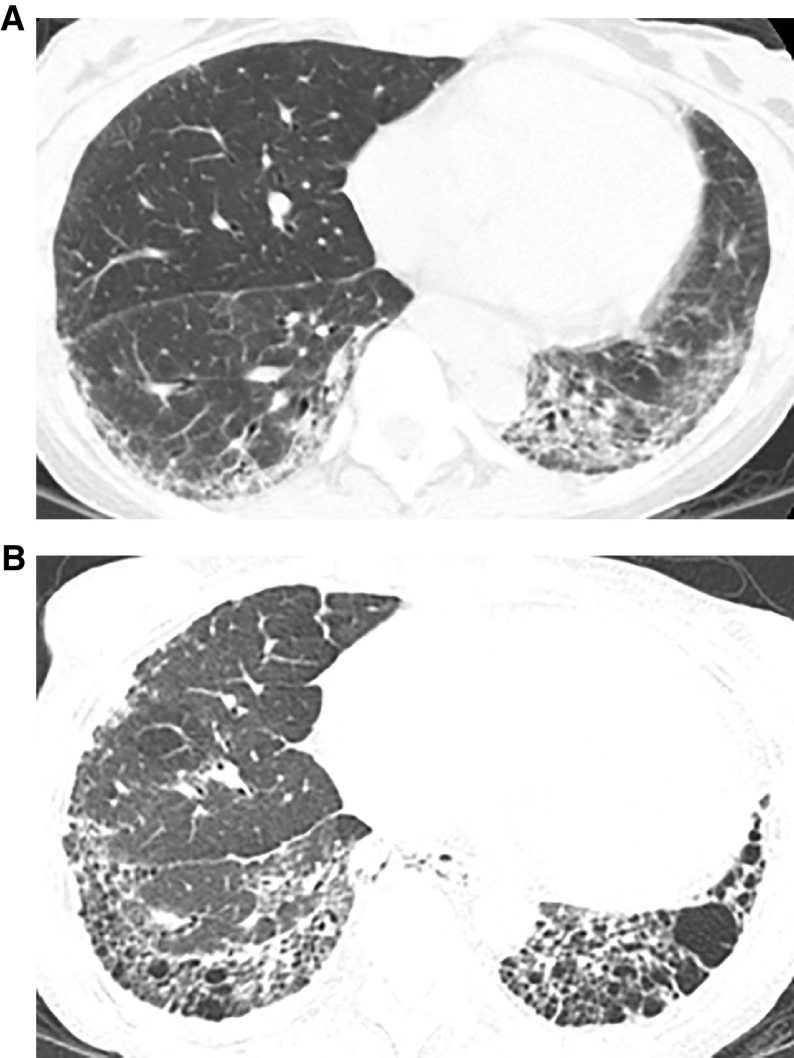
Comment in
-
Idiopathic Pulmonary Fibrosis Update: Reconciliation with Hypersensitivity Pneumonitis Guidelines Required?Am J Respir Crit Care Med. 2022 Nov 15;206(10):1293. doi: 10.1164/rccm.202205-0989LE. Am J Respir Crit Care Med. 2022. PMID: 35671476 Free PMC article. No abstract available.
-
Reply to Moran-Mendoza: Idiopathic Pulmonary Fibrosis Update. Reconciliation with Hypersensitivity Pneumonitis Guidelines Required?Am J Respir Crit Care Med. 2022 Nov 15;206(10):1298. doi: 10.1164/rccm.202206-1058LE. Am J Respir Crit Care Med. 2022. PMID: 35671482 Free PMC article. No abstract available.
-
Progressive Pulmonary Fibrosis: Should the Timelines Be Taken Out of the Definition?Am J Respir Crit Care Med. 2022 Nov 15;206(10):1293-1294. doi: 10.1164/rccm.202206-1143LE. Am J Respir Crit Care Med. 2022. PMID: 35868029 Free PMC article. No abstract available.
-
Comment on Idiopathic Pulmonary Fibrosis (an Update) and Progressive Pulmonary Fibrosis in Adults.Am J Respir Crit Care Med. 2022 Nov 15;206(10):1296. doi: 10.1164/rccm.202207-1260LE. Am J Respir Crit Care Med. 2022. PMID: 35868030 Free PMC article. No abstract available.
-
Progressive Pulmonary Fibrosis: Putting the Cart Before the Horse.Am J Respir Crit Care Med. 2022 Nov 15;206(10):1294-1295. doi: 10.1164/rccm.202206-1166LE. Am J Respir Crit Care Med. 2022. PMID: 35868031 Free PMC article. No abstract available.
-
Anti-acid Therapies in Idiopathic Pulmonary Fibrosis: Premature to Dismiss?Am J Respir Crit Care Med. 2022 Nov 15;206(10):1297. doi: 10.1164/rccm.202207-1420LE. Am J Respir Crit Care Med. 2022. PMID: 35921176 Free PMC article. No abstract available.
References
-
- Raghu G, Collard HR, Egan JJ, Martinez FJ, Behr J, Brown KK, et al. ATS/ERS/JRS/ALAT Committee on Idiopathic Pulmonary Fibrosis An official ATS/ERS/JRS/ALAT statement: idiopathic pulmonary fibrosis: evidence-based guidelines for diagnosis and management. Am J Respir Crit Care Med . 2011;183:788–824. - PMC - PubMed
-
- Raghu G, Remy-Jardin M, Myers JL, Richeldi L, Ryerson CJ, Lederer DJ, et al. American Thoracic Society, European Respiratory Society, Japanese Respiratory Society, and Latin American Thoracic Society Diagnosis of idiopathic pulmonary fibrosis: an official ATS/ERS/JRS/ALAT clinical practice guideline. Am J Respir Crit Care Med . 2018;198:e44–e68. - PubMed
-
- Raghu G, Rochwerg B, Zhang Y, Garcia CAC, Azuma A, Behr J, et al. American Thoracic Society European Respiratory society; Japanese Respiratory Society; Latin American Thoracic Association. An official ATS/ERS/JRS/ALAT clinical practice guideline: treatment of idiopathic pulmonary fibrosis. An update of the 2011 clinical practice guideline. Am J Respir Crit Care Med . 2015;192:e3–e19. - PubMed
-
- Flaherty KR, Wells AU, Cottin V, Devaraj A, Walsh SLF, Inoue Y, et al. INBUILD Trial Investigators Nintedanib in progressive fibrosing interstitial lung diseases. N Engl J Med . 2019;381:1718–1727. - PubMed
-
- Wells AU, Brown KK, Flaherty KR, Kolb M, Thannickal VJ, IPF Consensus Working Group What’s in a name? That which we call IPF, by any other name would act the same. Eur Respir J . 2018;51:1800692. - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
