Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation
- PMID: 35486677
- PMCID: PMC9332111
- DOI: 10.1126/sciimmunol.abm8161
Succinate dehydrogenase/complex II is critical for metabolic and epigenetic regulation of T cell proliferation and inflammation
Abstract
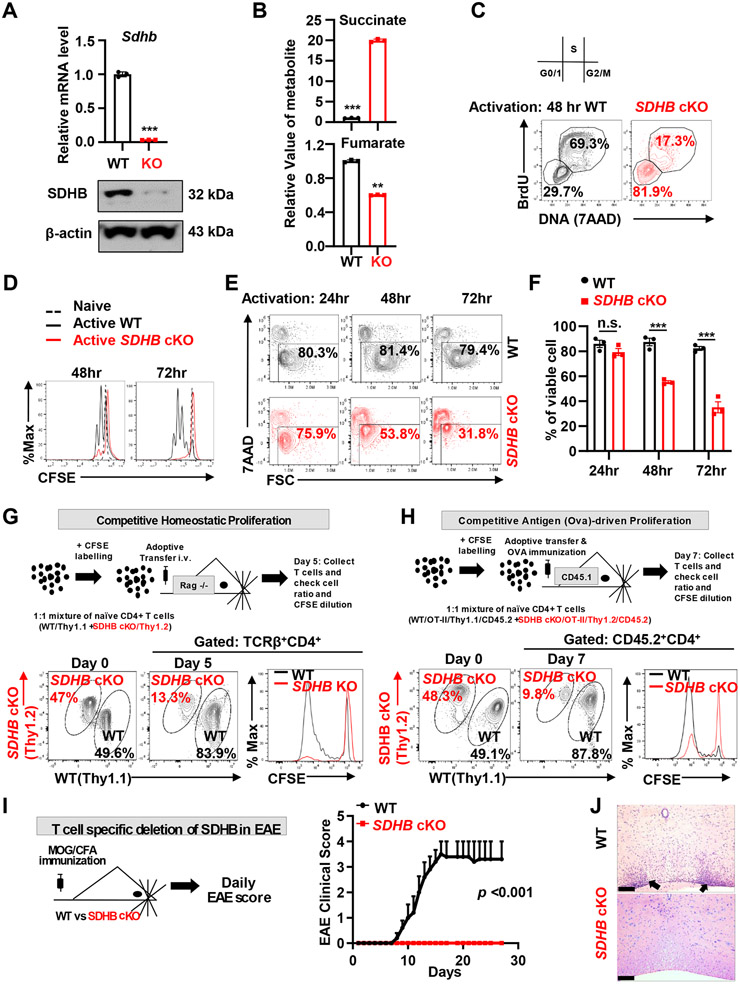
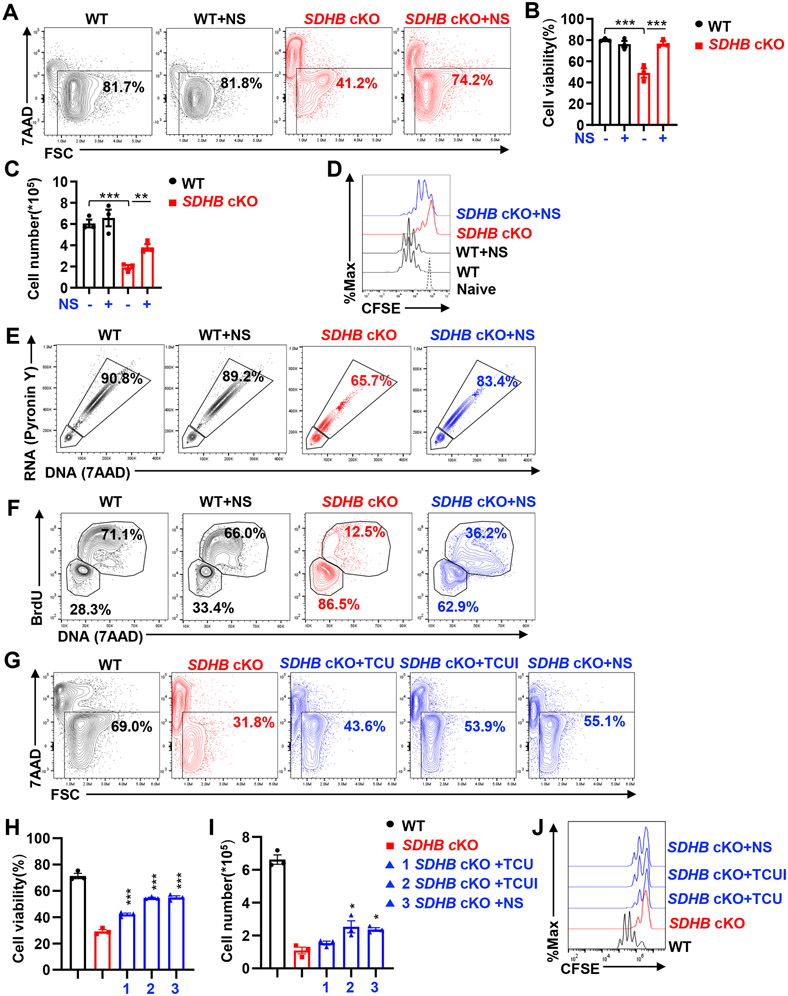
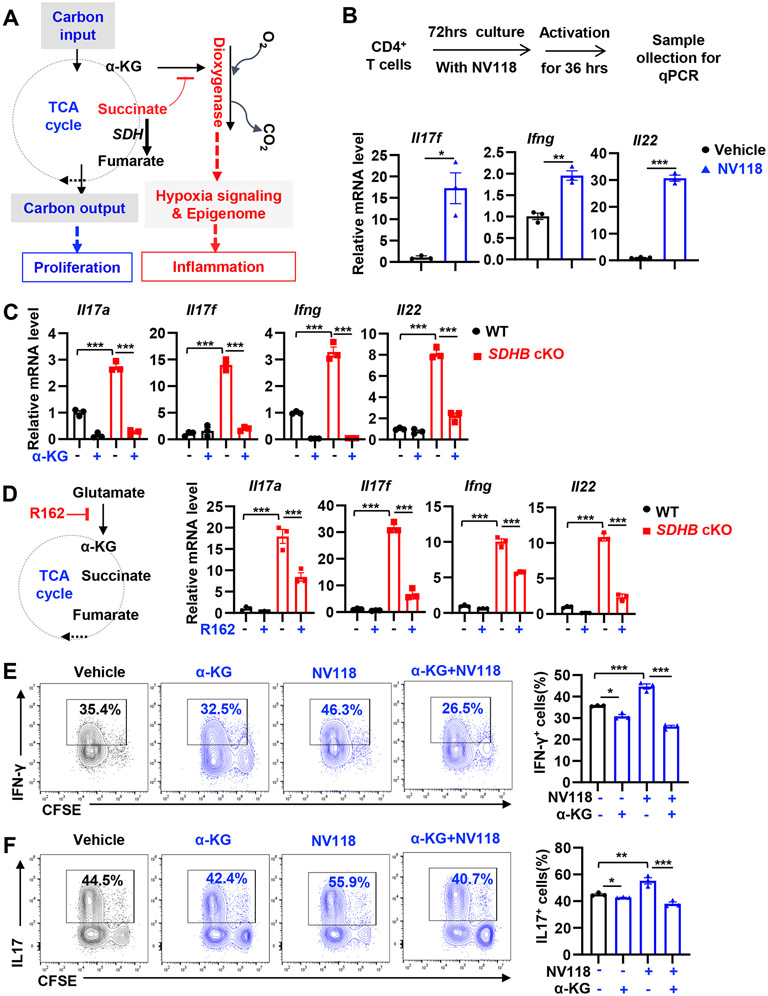
Effective T cell-mediated immune responses require the proper allocation of metabolic resources to sustain growth, proliferation, and cytokine production. Epigenetic control of the genome also governs T cell transcriptome and T cell lineage commitment and maintenance. Cellular metabolic programs interact with epigenetic regulation by providing substrates for covalent modifications of chromatin. By using complementary genetic, epigenetic, and metabolic approaches, we revealed that tricarboxylic acid (TCA) cycle flux fueled biosynthetic processes while controlling the ratio of succinate/α-ketoglutarate (α-KG) to modulate the activities of dioxygenases that are critical for driving T cell inflammation. In contrast to cancer cells, where succinate dehydrogenase (SDH)/complex II inactivation drives cell transformation and growth, SDH/complex II deficiency in T cells caused proliferation and survival defects when the TCA cycle was truncated, blocking carbon flux to support nucleoside biosynthesis. Replenishing the intracellular nucleoside pool partially relieved the dependence of T cells on SDH/complex II for proliferation and survival. SDH deficiency induced a proinflammatory gene signature in T cells and promoted T helper 1 and T helper 17 lineage differentiation. An increasing succinate/α-KG ratio in SDH-deficient T cells promoted inflammation by changing the pattern of the transcriptional and chromatin accessibility signatures and consequentially increasing the expression of the transcription factor, PR domain zinc finger protein 1. Collectively, our studies revealed a role of SDH/complex II in allocating carbon resources for anabolic processes and epigenetic regulation in T cell proliferation and inflammation.
Conflict of interest statement
Figures




References
Publication types
MeSH terms
Substances
Supplementary concepts
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials