

A novel β-catenin/BCL9 complex inhibitor blocks oncogenic Wnt signaling and disrupts cholesterol homeostasis in colorectal cancer
- PMID: 35486727
- PMCID: PMC9054024
- DOI: 10.1126/sciadv.abm3108
A novel β-catenin/BCL9 complex inhibitor blocks oncogenic Wnt signaling and disrupts cholesterol homeostasis in colorectal cancer
Abstract
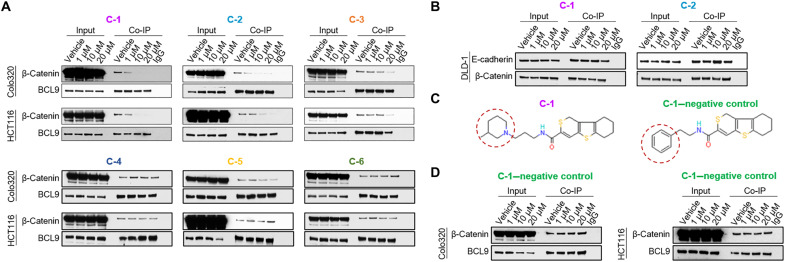
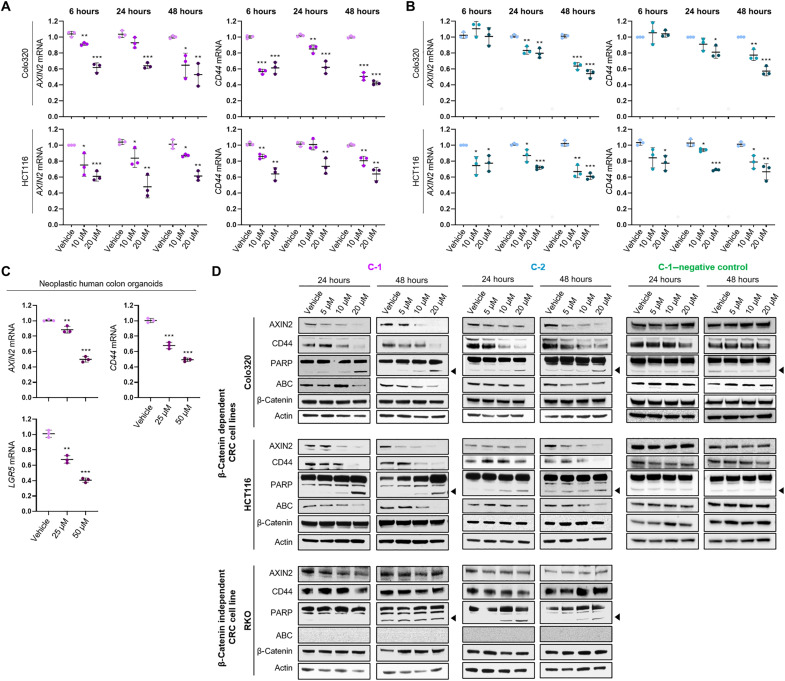
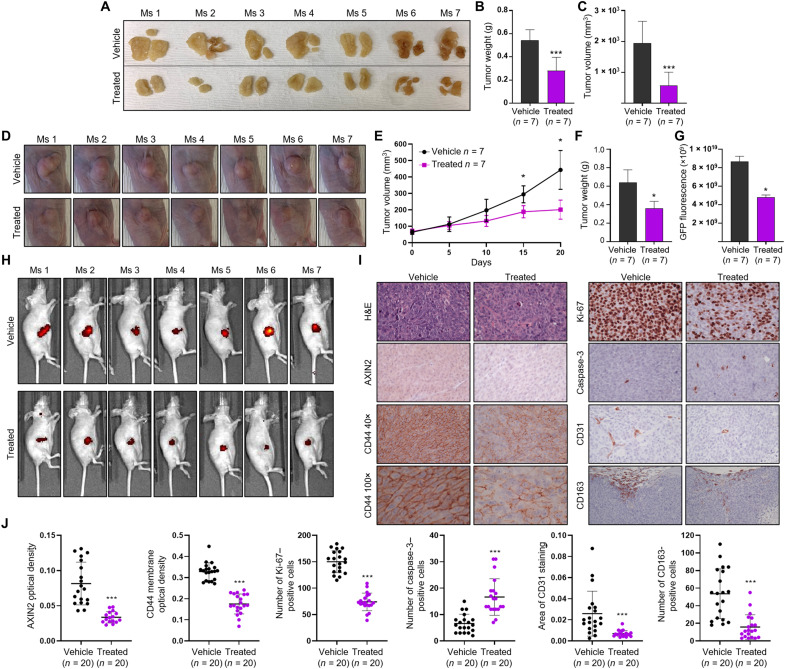
Dysregulated Wnt/β-catenin signaling is implicated in the pathogenesis of many human cancers, including colorectal cancer (CRC), making it an attractive clinical target. With the aim of inhibiting oncogenic Wnt activity, we developed a high-throughput screening AlphaScreen assay to identify selective small-molecule inhibitors of the interaction between β-catenin and its coactivator BCL9. We identified a compound that consistently bound to β-catenin and specifically inhibited in vivo native β-catenin/BCL9 complex formation in CRC cell lines. This compound inhibited Wnt activity, down-regulated expression of the Wnt/β-catenin signature in gene expression studies, disrupted cholesterol homeostasis, and significantly reduced the proliferation of CRC cell lines and tumor growth in a xenograft mouse model of CRC. This study has therefore identified a specific small-molecule inhibitor of oncogenic Wnt signaling, which may have value as a probe for functional studies and has important implications for the development of novel therapies in patients with CRC.
Figures





References
-
- Logan C. Y., Nusse R., The Wnt signaling pathway in development and disease. Annu. Rev. Cell Dev. Biol. 20, 781–810 (2004). - PubMed
-
- Steinhart Z., Angers S., Wnt signaling in development and tissue homeostasis. Development 145, dev146589 (2018). - PubMed
-
- Clevers H., Nusse R., Wnt/β-catenin signaling and disease. Cell 149, 1192–1205 (2012). - PubMed
-
- Klaus A., Birchmeier W., Wnt signalling and its impact on development and cancer. Nat. Rev. Cancer 8, 387–398 (2008). - PubMed
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
