

Incorporating local ancestry improves identification of ancestry-associated methylation signatures and meQTLs in African Americans
- PMID: 35488087
- PMCID: PMC9054854
- DOI: 10.1038/s42003-022-03353-5
Incorporating local ancestry improves identification of ancestry-associated methylation signatures and meQTLs in African Americans
Abstract
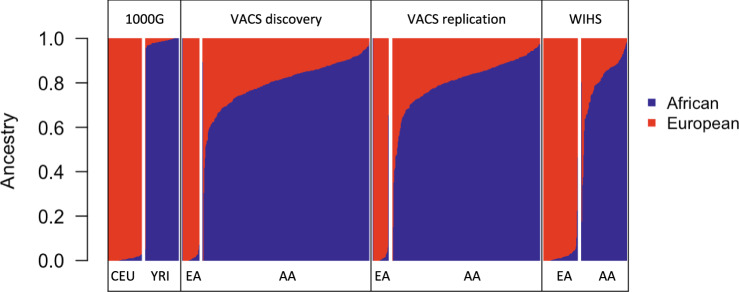
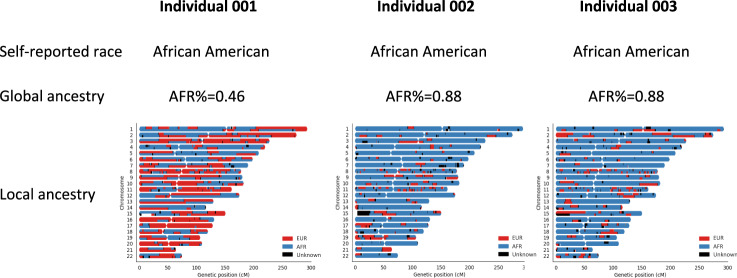
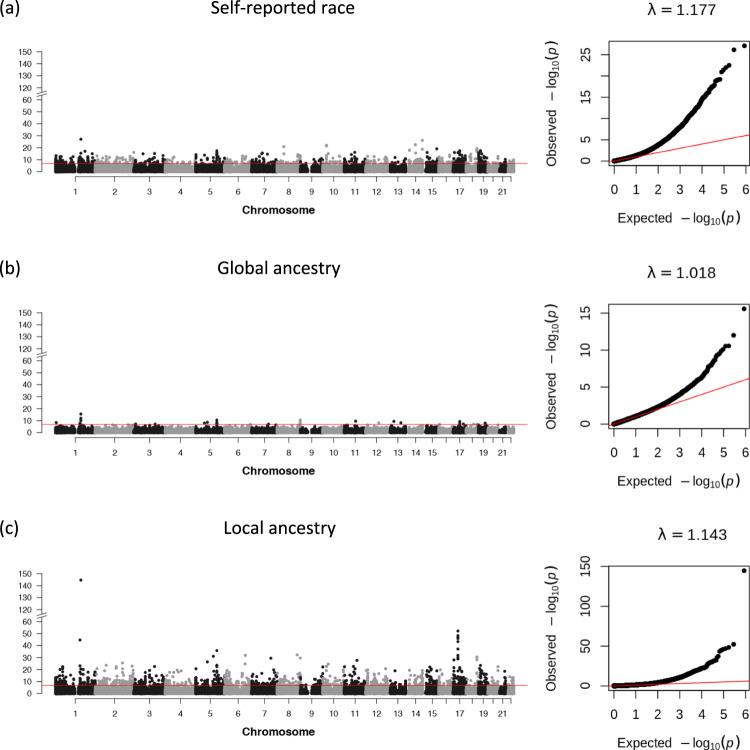
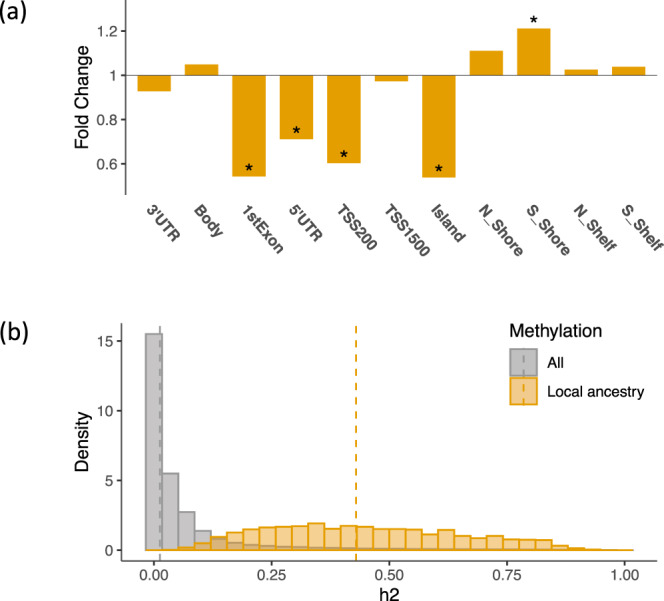
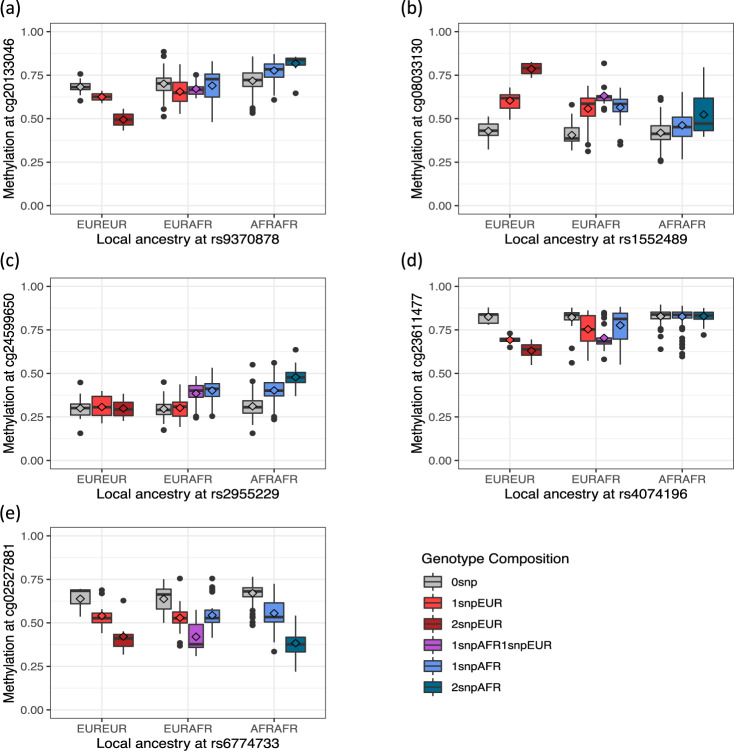
Here we report three epigenome-wide association studies (EWAS) of DNA methylation on self-reported race, global genetic ancestry, and local genetic ancestry in admixed Americans from three sets of samples, including internal and external replications (Ntotal = 1224). Our EWAS on local ancestry (LA) identified the largest number of ancestry-associated DNA methylation sites and also featured the highest replication rate. Furthermore, by incorporating ancestry origins of genetic variations, we identified 36 methylation quantitative trait loci (meQTL) clumps for LA-associated CpGs that cannot be captured by a model that assumes identical genetic effects across ancestry origins. Lead SNPs at 152 meQTL clumps had significantly different genetic effects in the context of an African or European ancestry background. Local ancestry information enables superior capture of ancestry-associated methylation signatures and identification of ancestry-specific genetic effects on DNA methylation. These findings highlight the importance of incorporating local ancestry for EWAS in admixed samples from multi-ancestry cohorts.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
Comment in
-
Genetic differentiation at probe SNPs leads to spurious results in meQTL discovery.Commun Biol. 2023 Dec 21;6(1):1295. doi: 10.1038/s42003-023-05658-5. Commun Biol. 2023. PMID: 38129663 Free PMC article. No abstract available.
References
-
- Xia Y-Y, et al. Racial/ethnic disparities in human DNA methylation. Biochimica Biophysica Acta. 2014;1846:258–262. - PubMed
Publication types
MeSH terms
Associated data
Grants and funding
- P30 AI050409/AI/NIAID NIH HHS/United States
- R01 DA047063/DA/NIDA NIH HHS/United States
- U01 HL146245/HL/NHLBI NIH HHS/United States
- U01 AA020795/AA/NIAAA NIH HHS/United States
- U01 HL146242/HL/NHLBI NIH HHS/United States
- U01 HL146193/HL/NHLBI NIH HHS/United States
- U01 AA020790/AA/NIAAA NIH HHS/United States
- U01 AA020799/AA/NIAAA NIH HHS/United States
- UL1 TR000004/TR/NCATS NIH HHS/United States
- R03 DA039745/DA/NIDA NIH HHS/United States
- U01 HL146241/HL/NHLBI NIH HHS/United States
- P30 AI027767/AI/NIAID NIH HHS/United States
- U01 HL146205/HL/NHLBI NIH HHS/United States
- R01 DA038632/DA/NIDA NIH HHS/United States
- R01 DA047820/DA/NIDA NIH HHS/United States
- U01 HL146192/HL/NHLBI NIH HHS/United States
- U10 AA013566/AA/NIAAA NIH HHS/United States
- R01 DA042691/DA/NIDA NIH HHS/United States
- U01 HL146204/HL/NHLBI NIH HHS/United States
- U01 HL146202/HL/NHLBI NIH HHS/United States
- U01 HL146194/HL/NHLBI NIH HHS/United States
- U24 AA020794/AA/NIAAA NIH HHS/United States
- R01 DA035616/DA/NIDA NIH HHS/United States
- U01 HL146203/HL/NHLBI NIH HHS/United States
- P30 AI050410/AI/NIAID NIH HHS/United States
LinkOut - more resources
Full Text Sources
