

Batch-effect detection, correction and characterisation in Illumina HumanMethylation450 and MethylationEPIC BeadChip array data
- PMID: 35488315
- PMCID: PMC9055778
- DOI: 10.1186/s13148-022-01277-9
Batch-effect detection, correction and characterisation in Illumina HumanMethylation450 and MethylationEPIC BeadChip array data
Abstract
Background: Genomic technologies can be subject to significant batch-effects which are known to reduce experimental power and to potentially create false positive results. The Illumina Infinium Methylation BeadChip is a popular technology choice for epigenome-wide association studies (EWAS), but presently, little is known about the nature of batch-effects on these designs. Given the subtlety of biological phenotypes in many EWAS, control for batch-effects should be a consideration.
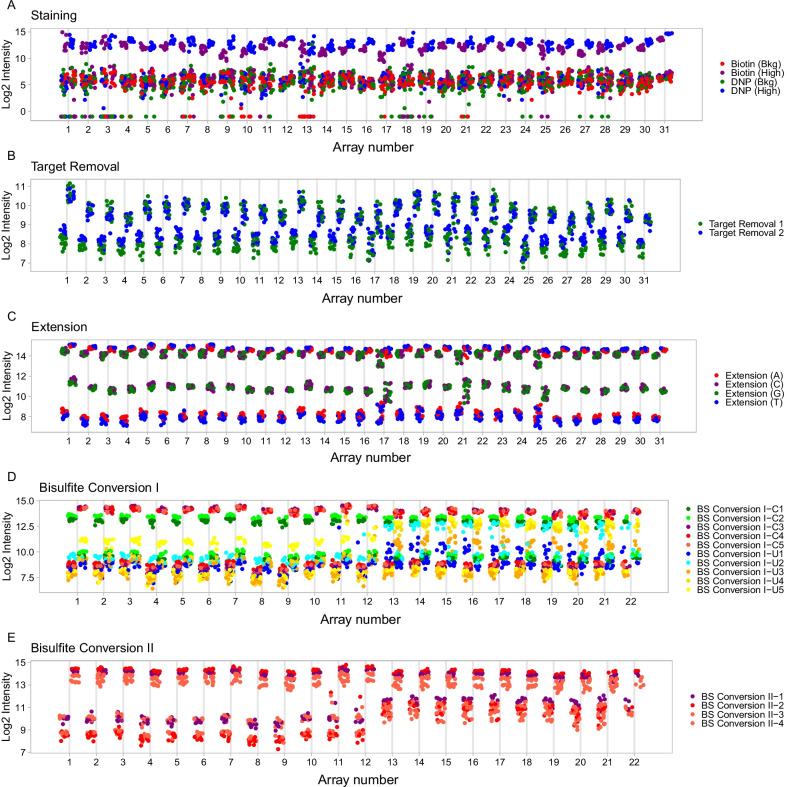
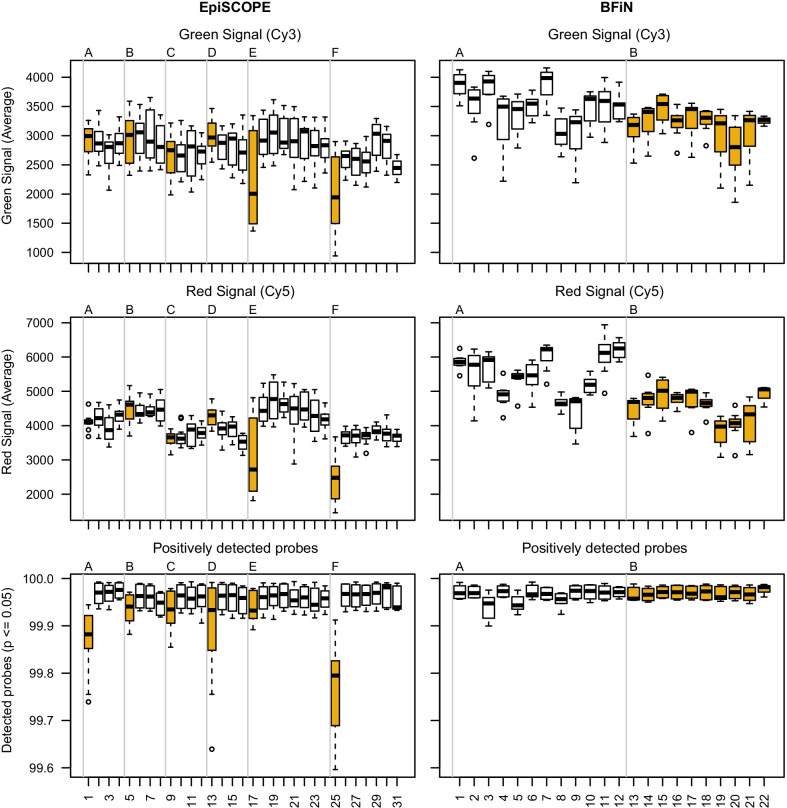
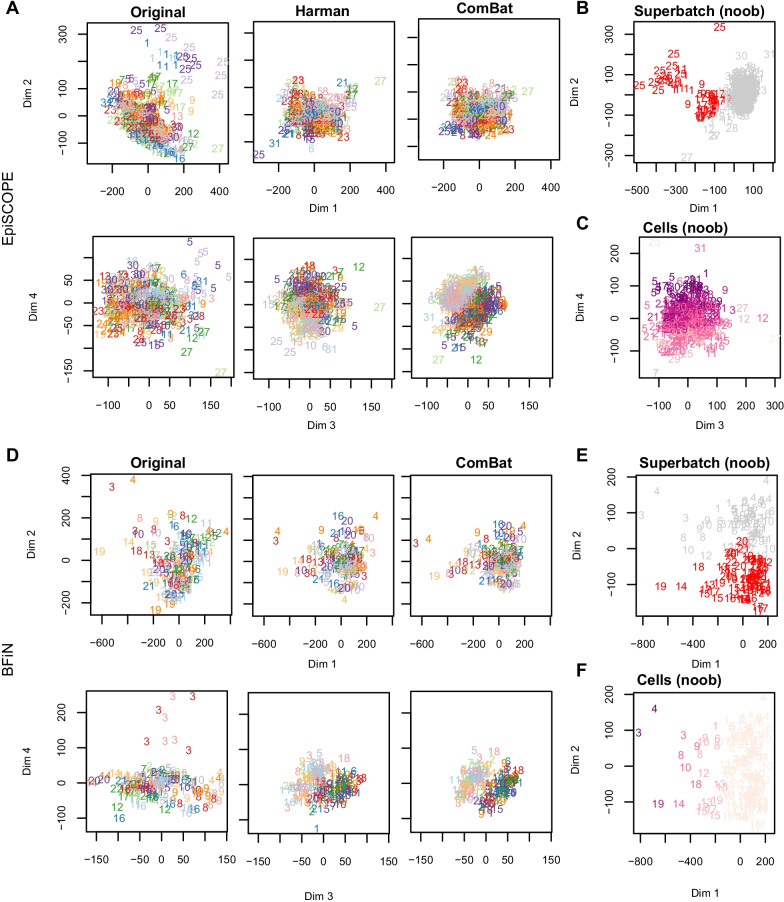
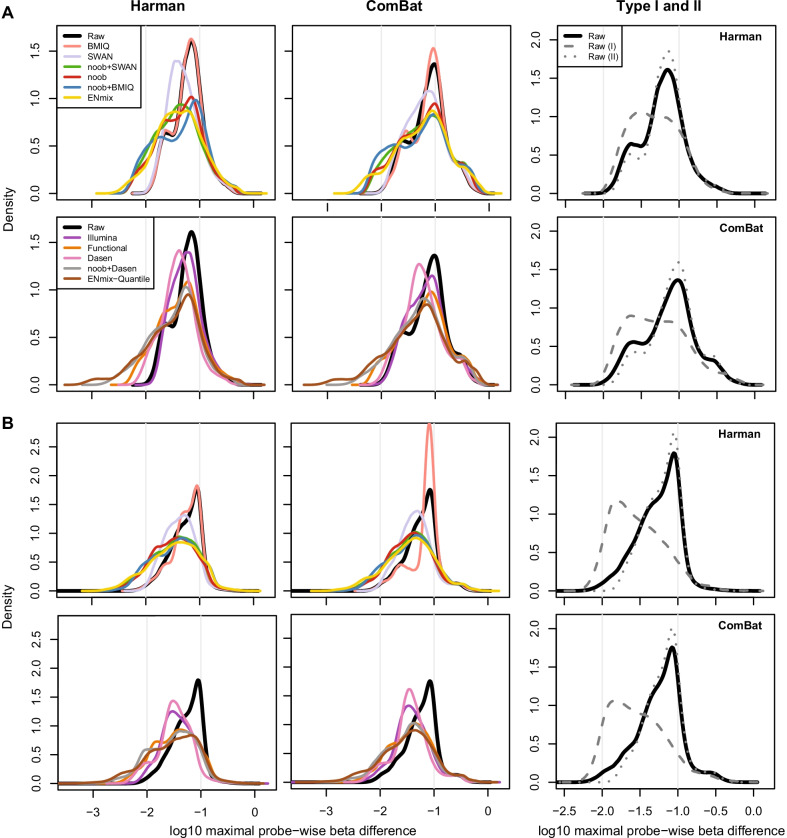
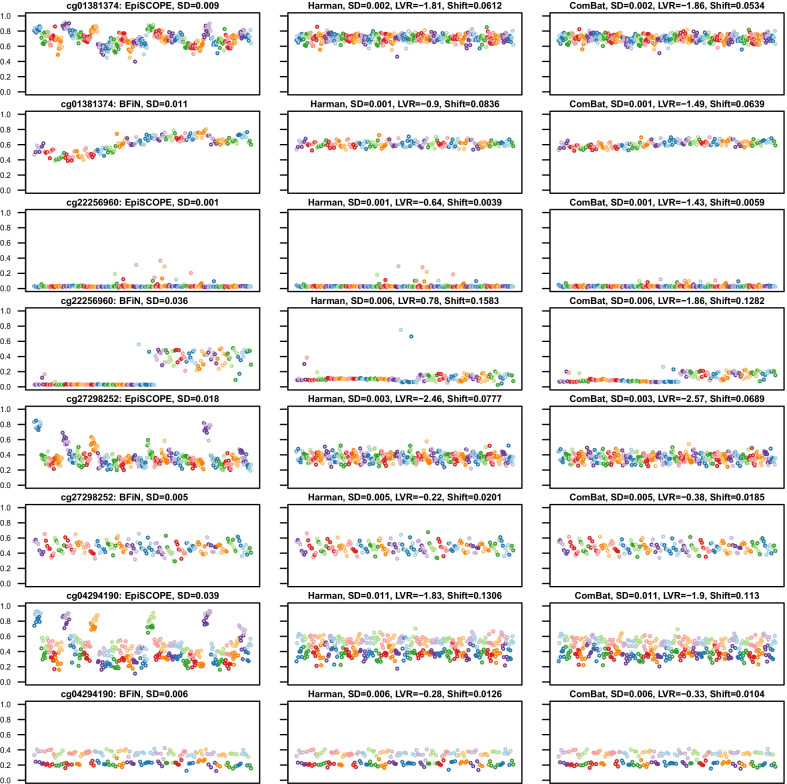
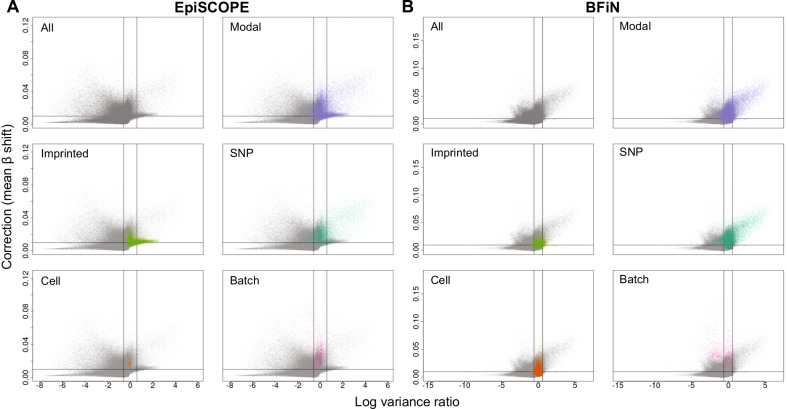
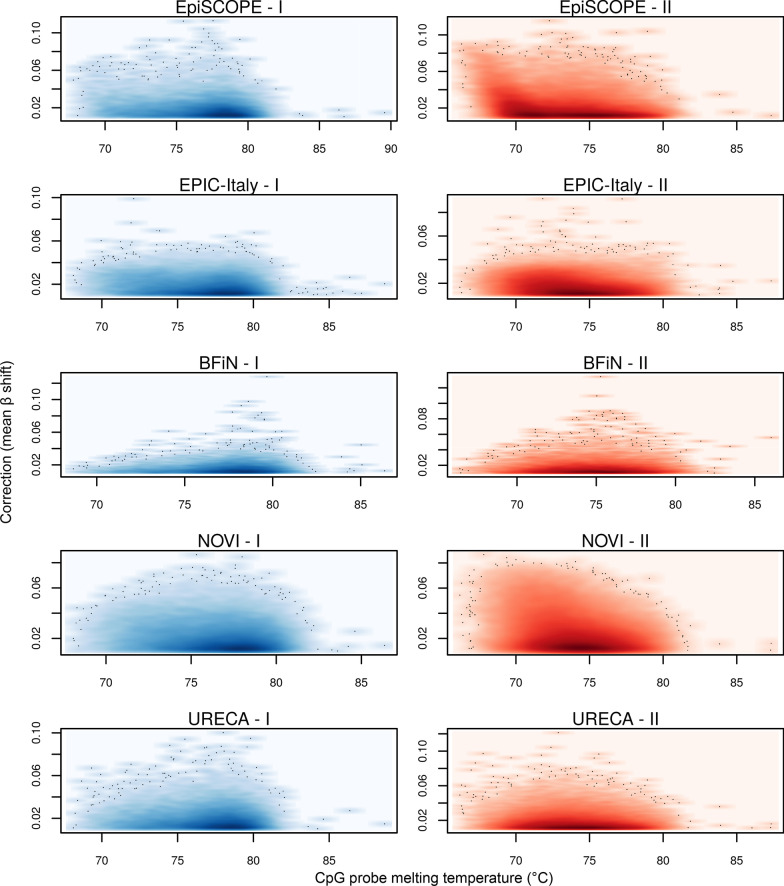
Results: Using the batch-effect removal approaches in the ComBat and Harman software, we examined two in-house datasets and compared results with three large publicly available datasets, (1214 HumanMethylation450 and 1094 MethylationEPIC BeadChips in total), and find that despite various forms of preprocessing, some batch-effects persist. This residual batch-effect is associated with the day of processing, the individual glass slide and the position of the array on the slide. Consistently across all datasets, 4649 probes required high amounts of correction. To understand the impact of this set to EWAS studies, we explored the literature and found three instances where persistently batch-effect prone probes have been reported in abstracts as key sites of differential methylation. As well as batch-effect susceptible probes, we also discover a set of probes which are erroneously corrected. We provide batch-effect workflows for Infinium Methylation data and provide reference matrices of batch-effect prone and erroneously corrected features across the five datasets spanning regionally diverse populations and three commonly collected biosamples (blood, buccal and saliva).
Conclusions: Batch-effects are ever present, even in high-quality data, and a strategy to deal with them should be part of experimental design, particularly for EWAS. Batch-effect removal tools are useful to reduce technical variance in Infinium Methylation data, but they need to be applied with care and make use of post hoc diagnostic measures.
Keywords: Batch-effect; Clustering; ComBat; EWAS; False positives; Harman; Infinium; Methylation; SNP.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures



References
-
- Luo J, Schumacher M, Scherer A, Sanoudou D, Megherbi D, Davison T, Shi T, Tong W, Shi L, Hong H, et al. A comparison of batch effect removal methods for enhancement of prediction performance using MAQC-II microarray gene expression data. Pharmacogenomics J. 2010;10:278–291. doi: 10.1038/tpj.2010.57. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
