

International consensus on diagnosis and management of Dravet syndrome
- PMID: 35490361
- PMCID: PMC9543220
- DOI: 10.1111/epi.17274
International consensus on diagnosis and management of Dravet syndrome
Abstract
Objective: This study was undertaken to gain consensus from experienced physicians and caregivers regarding optimal diagnosis and management of Dravet syndrome (DS), in the context of recently approved, DS-specific therapies and emerging disease-modifying treatments.
Methods: A core working group was convened consisting of six physicians with recognized expertise in DS and two representatives of the Dravet Syndrome Foundation. This core group summarized the current literature (focused on clinical presentation, comorbidities, maintenance and rescue therapies, and evolving disease-modifying therapies) and nominated the 31-member expert panel (ensuring international representation), which participated in two rounds of a Delphi process to gain consensus on diagnosis and management of DS.
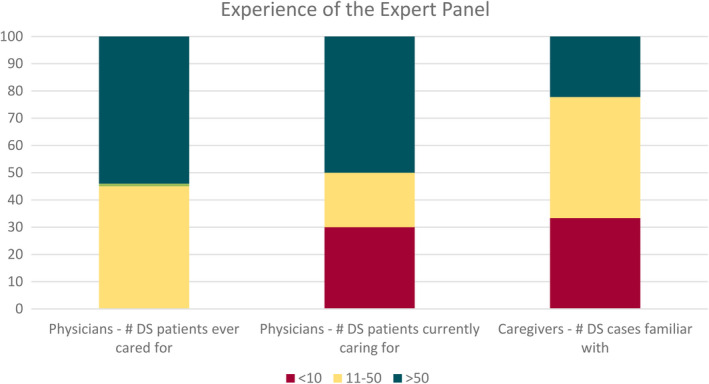
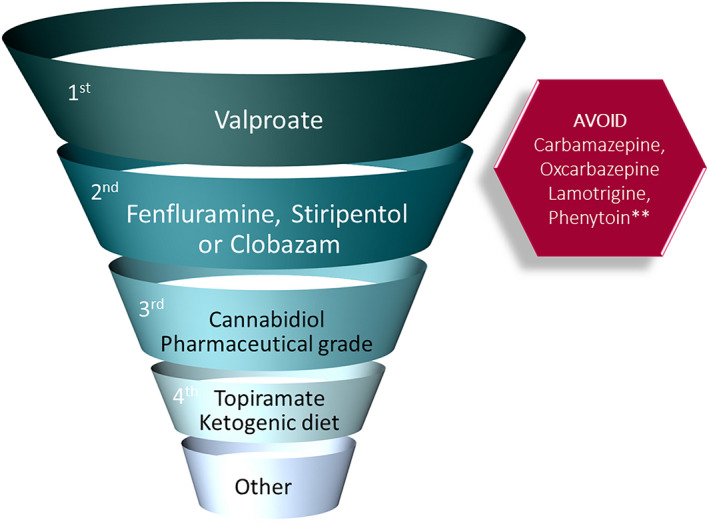
Results: There was strong consensus that infants 2-15 months old, presenting with either a first prolonged hemiclonic seizure or first convulsive status epilepticus with fever or following vaccination, in the absence of another cause, should undergo genetic testing for DS. Panelists agreed on evolution of specific comorbidities with time, but less agreement was achieved on optimal management. There was also agreement on appropriate first- to third-line maintenance therapies, which included the newly approved agents. Whereas there was agreement for recommendation of disease-modifying therapies, if they are proven safe and efficacious for seizures and/or reduction of comorbidities, there was less consensus for when these should be started, with caregivers being more conservative than physicians.
Significance: This International DS Consensus, informed by both experienced global caregiver and physician voices, provides a strong overview of the impact of DS, therapeutic goals and optimal management strategies incorporating the recent therapeutic advances in DS, and evolving disease-modifying therapies.
Keywords: SCN1A; cannabidiol; developmental and epileptic encephalopathy; disease-modifying treatment; fenfluramine; stiripentol.
© 2022 The Authors. Epilepsia published by Wiley Periodicals LLC on behalf of International League Against Epilepsy.
Conflict of interest statement
E.C.W. has served as a paid consultant for Encoded Therapeutics, Eisai, Epygenix, and BioMarin. She is Editor‐in‐Chief of Epilepsy.com. K.G.K. has received research funding from Zogenix, Encoded, Eisai, and West Pharmaceuticals. She has participated on data and safety monitoring boards for GW Pharmaceuticals and Epygenix, and has received consulting funds from BioMarin, Zogenix, Encoded, Eisai, Stoke, and Biocodex. R.N. has served as principal investigator in clinical trials for Novartis, Nutricia, Eisai, UCB, GW Pharma, and LivaNova. She has received consulting and lecturer honoraria from Biogene, BioMarin, Praxis, GW Pharma, Zogenix, Novartis, Nutricia, Stoke, Ionis, Targeon, Neuraxpharma, Takeda, Nutricia, Biocodex, Advicennes, and Eisai. She has received unrestricted research grants from Eisai, UCB, LivaNova, and GW Pharma and academic research grants from EJP‐RD (Horizons 2020). I.S. has served on scientific advisory boards for UCB, Eisai, GlaxoSmithKline, BioMarin, Nutricia, Rogcon, Chiesi, Encoded Therapeutics, Knopp Biosciences, and Xenon Pharmaceuticals; has received speaker honoraria from GlaxoSmithKline, UCB, BioMarin, Biocodex, Chiesi, LivaNova, and Eisai; has received funding for travel from UCB, Biocodex, GlaxoSmithKline, BioMarin, and Eisai; has served as an investigator for Zogenix, Zynerba, Ultragenyx, GW Pharma, UCB, Eisai, Xenon Pharmaceuticals, Anavex Life Sciences, Ovid Therapeutics, Epygenix, Encoded Therapeutics, and Marinus; has consulted for Zynerba Pharmaceuticals, Atheneum Partners, Ovid Therapeutics, Care Beyond Diagnosis, Epilepsy Consortium, and UCB; and is a Non‐Executive Director of Bellberry. She may accrue future revenue on pending patent WO61/010176 (filed 2008): Therapeutic Compound; has a patent for
Figures
References
-
- Wirrell EC, Laux L, Donner E, Jette N, Knupp K, Meskis MA, et al. Optimizing the diagnosis and management of Dravet syndrome: recommendations from a North American consensus panel. Pediatr Neurol. 2017;68:18–34.e3. - PubMed
-
- Cross JH, Caraballo RH, Nabbout R, Vigevano F, Guerrini R, Lagae L. Dravet syndrome: treatment options and management of prolonged seizures. Epilepsia. 2019;60(Suppl 3):S39–48. - PubMed
-
- Devinsky O, Cross JH, Laux L, Marsh E, Miller I, Nabbout R, et al. Trial of cannabidiol for drug‐resistant seizures in the Dravet syndrome. N Engl J Med. 2017;376:2011–20. - PubMed
-
- Lagae L, Sullivan J, Knupp K, Laux L, Polster T, Nikanorova M, et al. Fenfluramine hydrochloride for the treatment of seizures in Dravet syndrome: a randomised, double‐blind, placebo‐controlled trial. Lancet. 2019;394:2243–54. - PubMed
Publication types
MeSH terms
Supplementary concepts
LinkOut - more resources
Full Text Sources
