

Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics
- PMID: 35491624
- PMCID: PMC9306285
- DOI: 10.1158/2159-8290.CD-21-1334
Adapted to Survive: Targeting Cancer Cells with BH3 Mimetics
Abstract
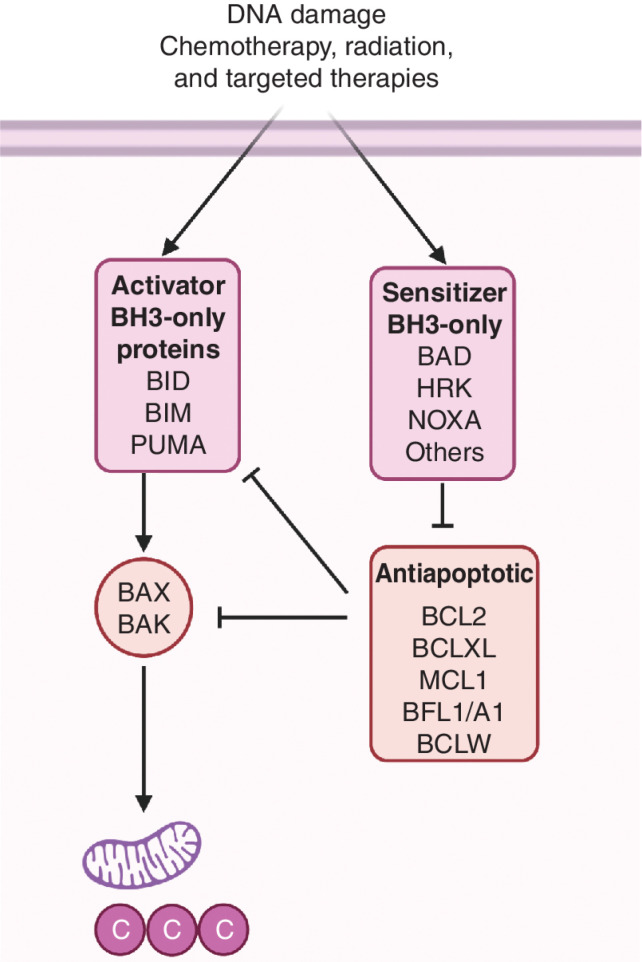
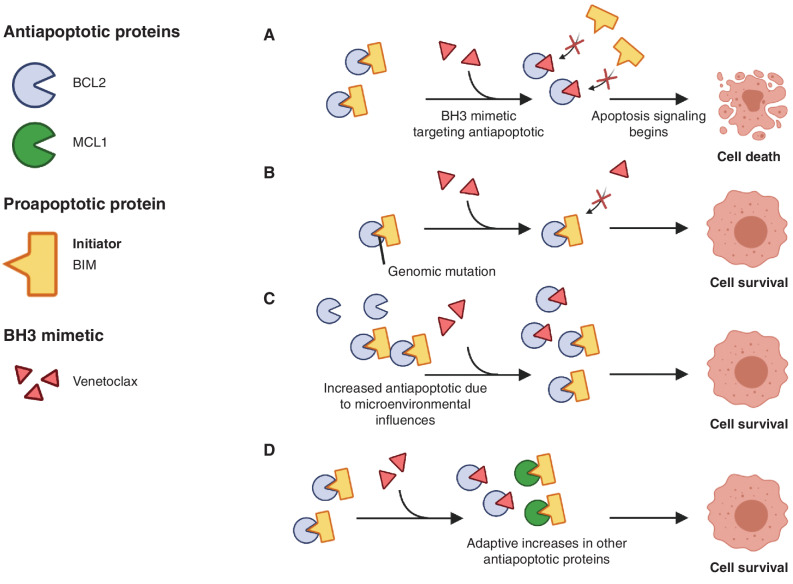
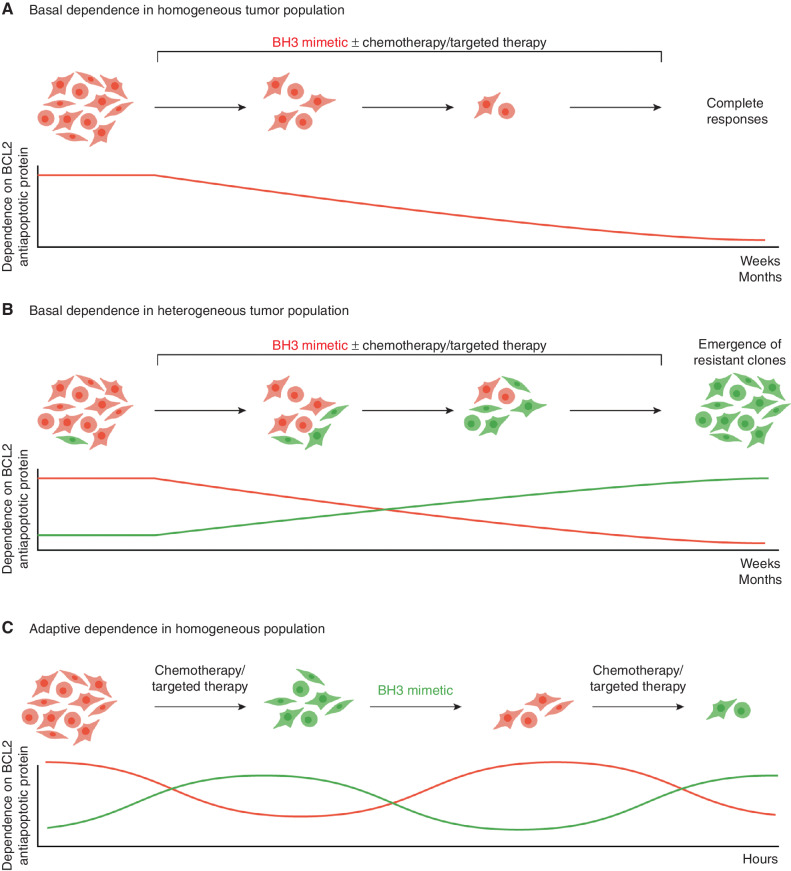
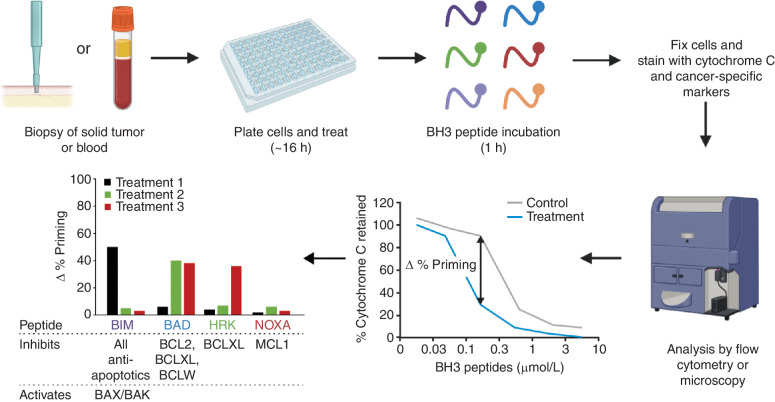
A hallmark of cancer is cell death evasion, underlying suboptimal responses to chemotherapy, targeted agents, and immunotherapies. The approval of the antiapoptotic BCL2 antagonist venetoclax has finally validated the potential of targeting apoptotic pathways in patients with cancer. Nevertheless, pharmacologic modulators of cell death have shown markedly varied responses in preclinical and clinical studies. Here, we review emerging concepts in the use of this class of therapies. Building on these observations, we propose that treatment-induced changes in apoptotic dependency, rather than pretreatment dependencies, will need to be recognized and targeted to realize the precise deployment of these new pharmacologic agents.
Significance: Targeting antiapoptotic family members has proven efficacious and tolerable in some cancers, but responses are infrequent, particularly for patients with solid tumors. Biomarkers to aid patient selection have been lacking. Precision functional approaches that overcome adaptive resistance to these compounds could drive durable responses to chemotherapy, targeted therapy, and immunotherapies.
©2022 The Authors; Published by the American Association for Cancer Research.
Figures
References
-
- Degterev A, Huang Z, Boyce M, Li Y, Jagtap P, Mizushima Net al. Chemical inhibitor of nonapoptotic cell death with therapeutic potential for ischemic brain injury. Nat Chem Biol 2005;1:112–9. - PubMed
-
- Kaczmarek A, Vandenabeele P, Krysko DV. Necroptosis: the release of damage-associated molecular patterns and its physiological relevance. Immunity 2013;38:209–23. - PubMed
-
- Izzo V, Bravo-San Pedro JM, Sica V, Kroemer G, Galluzzi L. Mitochondrial permeability transition: new findings and persisting uncertainties. Trends Cell Biol 2016;26:655–67. - PubMed
