

Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis
- PMID: 35491858
- PMCID: PMC9809951
- DOI: 10.1080/15548627.2022.2072054
Tubular cells produce FGF2 via autophagy after acute kidney injury leading to fibroblast activation and renal fibrosis
Abstract
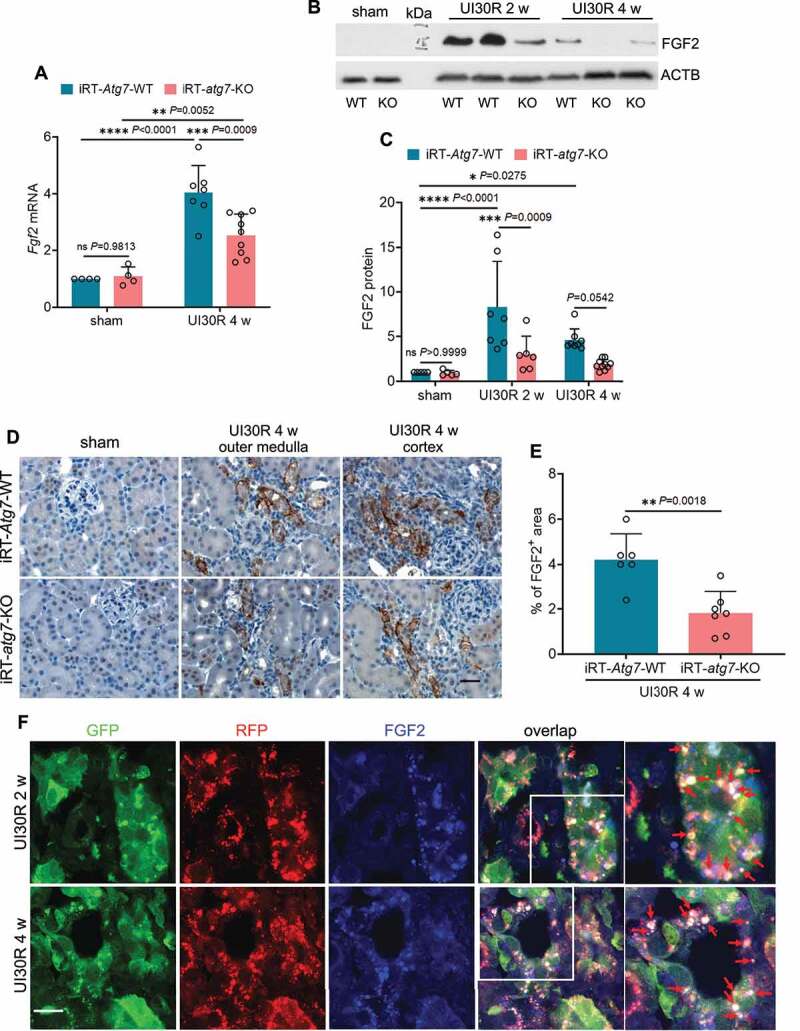
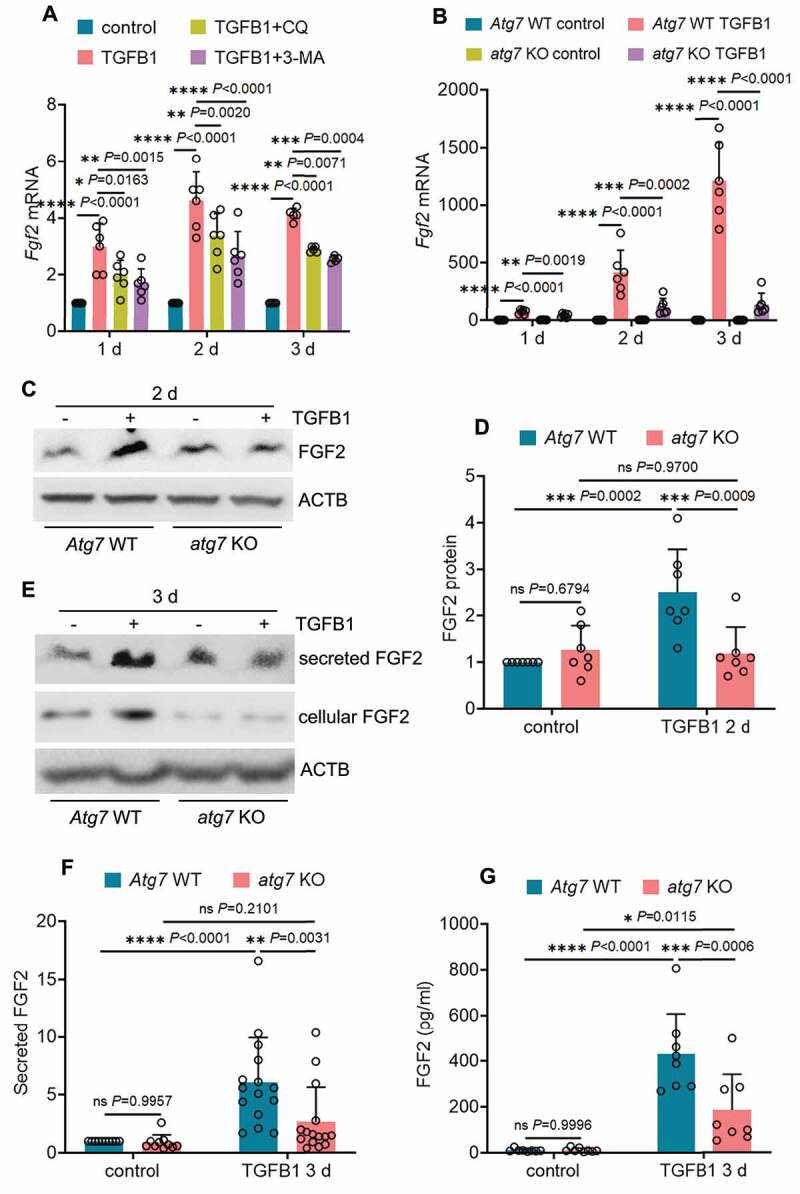
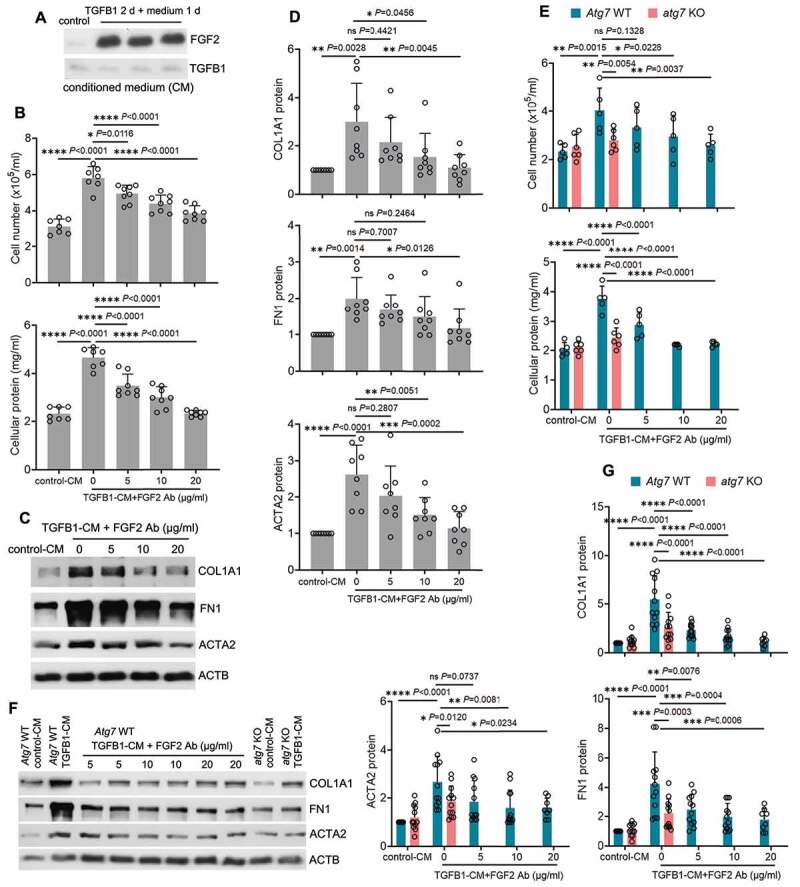
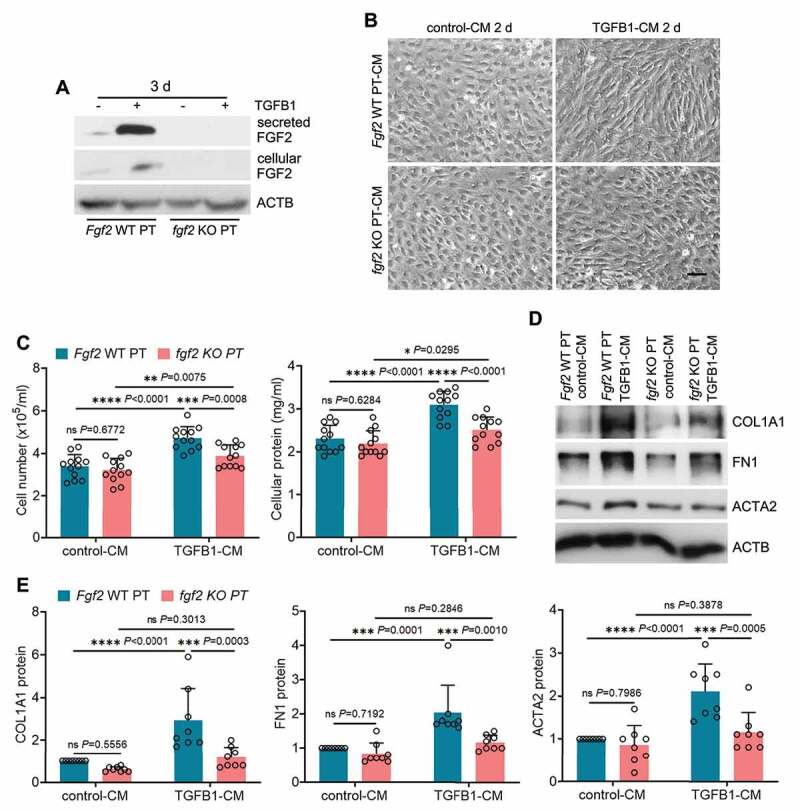
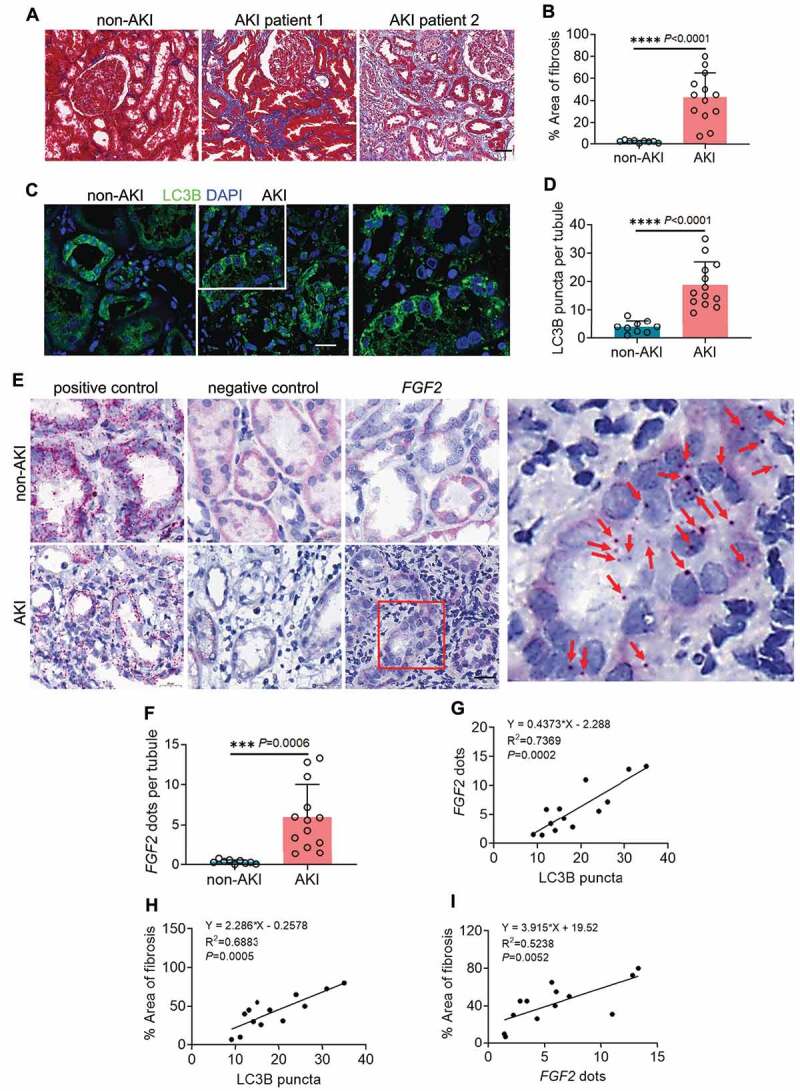
Following acute kidney injury (AKI), renal tubular cells may stimulate fibroblasts in a paracrine fashion leading to interstitial fibrosis, but the paracrine factors and their regulation under this condition remain elusive. Here we identify a macroautophagy/autophagy-dependent FGF2 (fibroblast growth factor 2) production in tubular cells. Upon induction, FGF2 acts as a key paracrine factor to activate fibroblasts for renal fibrosis. After ischemic AKI in mice, autophagy activation persisted for weeks in renal tubular cells. In inducible, renal tubule-specific atg7 (autophagy related 7) knockout (iRT-atg7-KO) mice, autophagy deficiency induced after AKI suppressed the pro-fibrotic phenotype in tubular cells and reduced fibrosis. Among the major cytokines, tubular autophagy deficiency in iRT-atg7-KO mice specifically diminished FGF2. Autophagy inhibition also attenuated FGF2 expression in TGFB1/TGF-β1 (transforming growth factor, beta 1)-treated renal tubular cells. Consistent with a paracrine action, the culture medium of TGFB1-treated tubular cells stimulated renal fibroblasts, and this effect was suppressed by FGF2 neutralizing antibody and also by fgf2- or atg7-deletion in tubular cells. In human, compared with non-AKI, the renal biopsies from post-AKI patients had higher levels of autophagy and FGF2 in tubular cells, which showed significant correlations with renal fibrosis. These results indicate that persistent autophagy after AKI induces pro-fibrotic phenotype transformation in tubular cells leading to the expression and secretion of FGF2, which activates fibroblasts for renal fibrosis during maladaptive kidney repair.Abbreviations: 3-MA: 3-methyladnine; ACTA2/α-SMA: actin alpha 2, smooth muscle, aorta; ACTB/β-actin: actin, beta; AKI: acute kidney injury; ATG/Atg: autophagy related; BUN: blood urea nitrogen; CCN2/CTGF: cellular communication network factor 2; CDKN2A/p16: cyclin dependent kinase inhibitor 2A; CKD: chronic kidney disease; CM: conditioned medium; COL1A1: collagen, type I, alpha 1; COL4A1: collagen, type IV, alpha 1; CQ: chloroquine; ECM: extracellular matrix; eGFR: estimated glomerular filtration rate; ELISA: enzyme-linked immunosorbent assay; FGF2: fibroblast growth factor 2; FN1: fibronectin 1; FOXO3: forkhead box O3; GAPDH: glyceraldehyde-3-phosphate dehydrogenase; HAVCR1/KIM-1: hepatitis A virus cellular receptor 1; IHC: immunohistochemistry; IRI: ischemia-reperfusion injury; ISH: in situ hybridization; LTL: lotus tetragonolobus lectin; MAP1LC3B/LC3B: microtubule-associated protein 1 light chain 3 beta; MTOR: mechanistic target of rapamycin kinase; PDGFB: platelet derived growth factor, B polypeptide; PPIB/cyclophilin B: peptidylprolyl isomerase B; RT-qPCR: real time-quantitative PCR; SA-GLB1/β-gal: senescence-associated galactosidase, beta 1; SASP: senescence-associated secretory phenotype; sCr: serum creatinine; SQSTM1/p62: sequestosome 1; TASCC: TOR-autophagy spatial coupling compartment; TGFB1/TGF-β1: transforming growth factor, beta 1; VIM: vimentin.
Keywords: Autophagy; FGF2; interstitial fibrosis; kidney repair; proximal tubule; renal ischemia-reperfusion.
Conflict of interest statement
All authors declare that they have no conflict of interest.
Figures
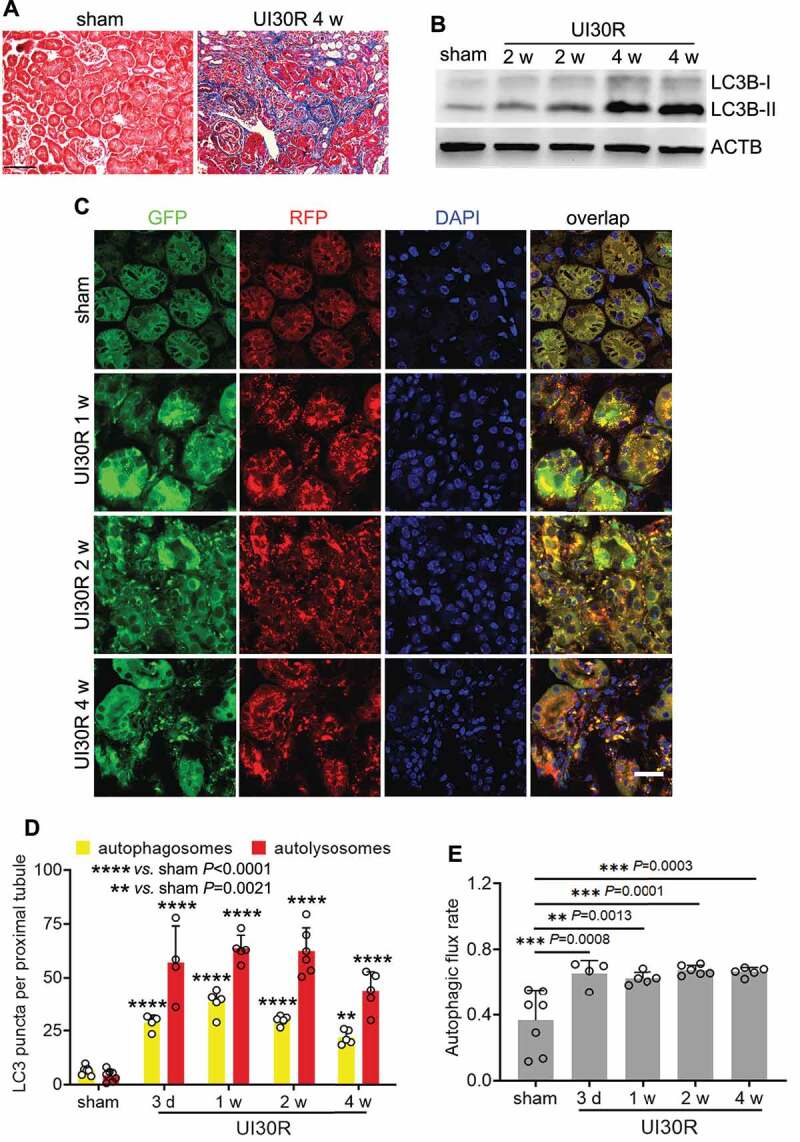
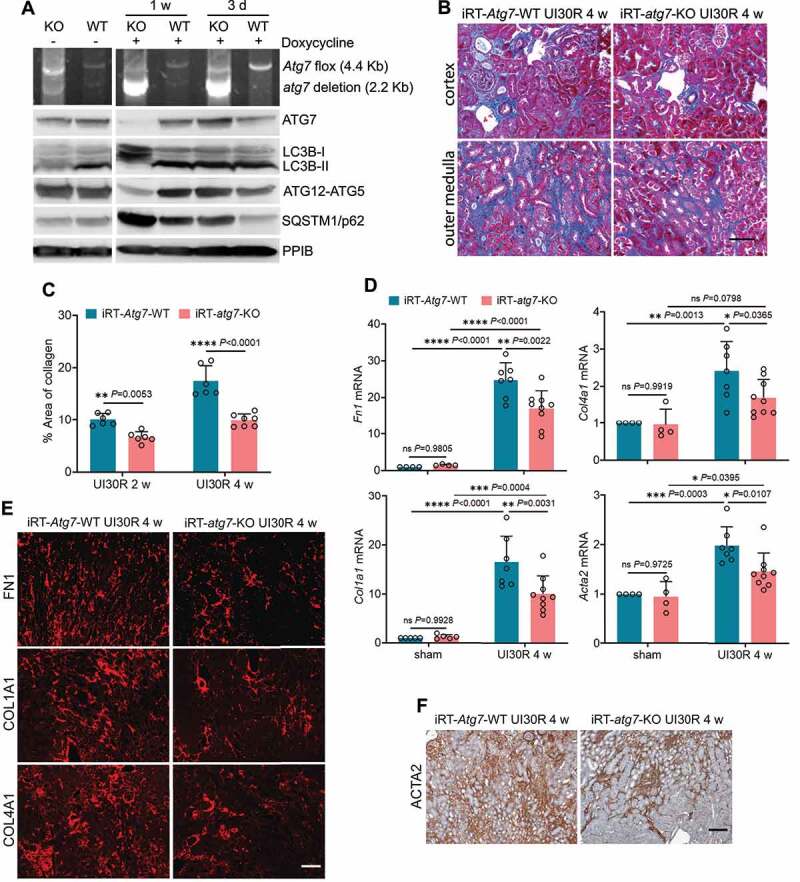
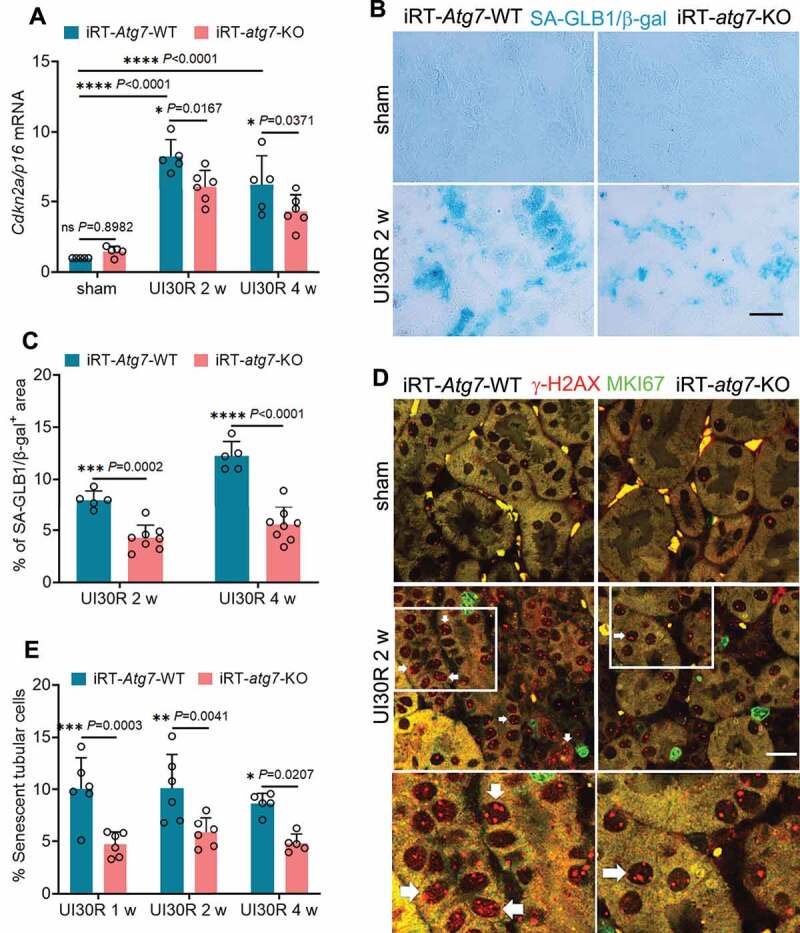
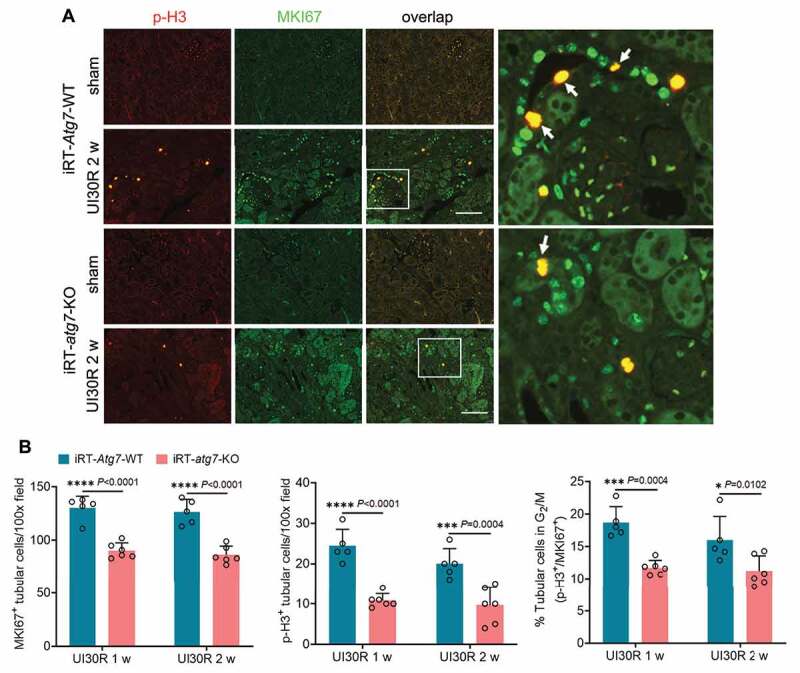
References
-
- Ronco C, Bellomo R, Kellum JA.. Acute kidney injury. Lancet. 2019;394:1949–1964. - PubMed
-
- Mehta RL, Burdmann EA, Cerda J, et al. Recognition and management of acute kidney injury in the International Society of Nephrology 0by25 global snapshot: a multinational cross-sectional study. Lancet. 2016;387:2017–2025. - PubMed
-
- See EJ, Jayasinghe K, Glassford N, et al. Long-term risk of adverse outcomes after acute kidney injury: a systematic review and meta-analysis of cohort studies using consensus definitions of exposure. Kidney Int. 2019;95:160–172. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials
Miscellaneous