

New Insights Into Pathophysiology of β-Thalassemia
- PMID: 35492364
- PMCID: PMC9041707
- DOI: 10.3389/fmed.2022.880752
New Insights Into Pathophysiology of β-Thalassemia
Abstract
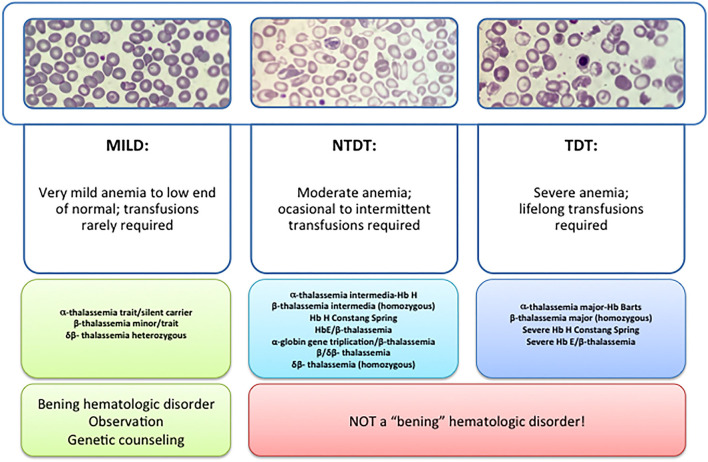
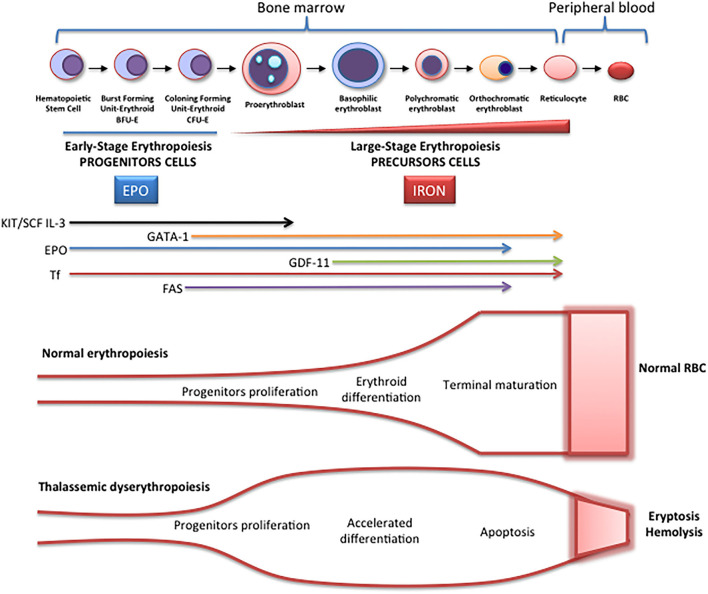
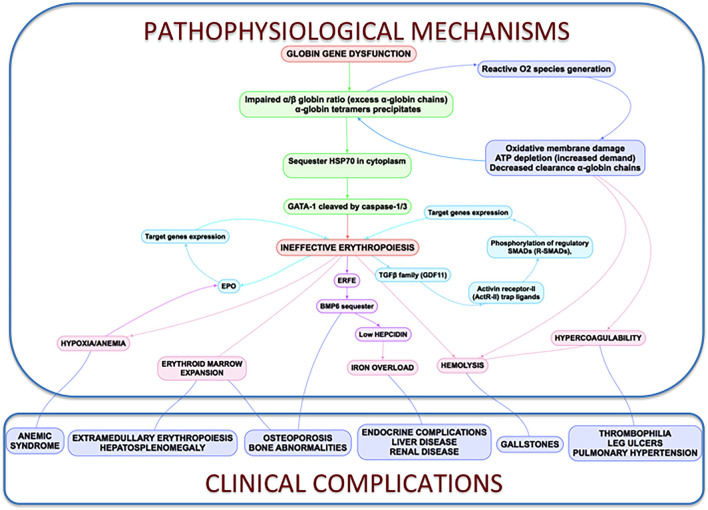
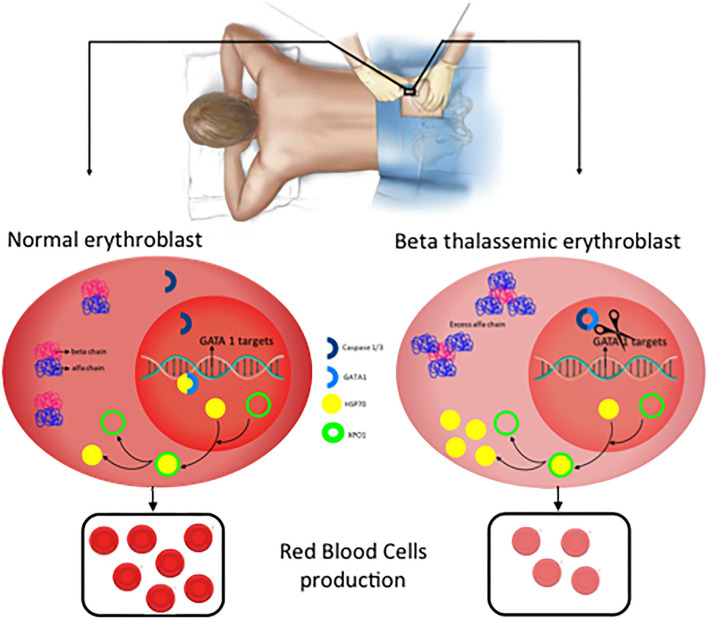
β-thalassemia is a disease caused by genetic mutations including a nucleotide change, small insertions or deletions in the β-globin gene, or in rare cases, gross deletions into the β-globin gene. These mutations affect globin-chain subunits within the hemoglobin tetramer what induces an imbalance in the α/β-globin chain ratio, with an excess of free α-globin chains that triggers the most important pathogenic events of the disease: ineffective erythropoiesis, chronic anemia/chronic hypoxia, compensatory hemopoietic expansion and iron overload. Based on advances in our knowledge of the pathophysiology of β-thalassemia, in recent years, emerging therapies and clinical trials are being conducted and are classified into three major categories based on the different approach features of the underlying pathophysiology: correction of the α/β-globin disregulation; improving iron overload and reverse ineffective erythropoiesis. However, pathways such as the dysregulation of transcriptional factors, activation of the inflammasome, or approach to mechanisms of bone mineral loss, remain unexplored for future therapeutic targets. In this review, we update the main pathophysiological pathways involved in β-thalassemia, focusing on the development of new therapies directed at new therapeutic targets.
Keywords: GATA1; anemia; inflammasome; thalassemia; β-globin.
Copyright © 2022 Sanchez-Villalobos, Blanquer, Moraleda, Salido and Perez-Oliva.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Cappellini MD, Cohen A, Porter J, Taher A, Viprakasit V. (eds.). Guidelines for the Management of Transfusion Dependent Thalassaemia (TDT). Nicosia: Thalassaemia International Federation; (2014). - PubMed
Publication types
LinkOut - more resources
Full Text Sources