

Cross-species genetic screens identify transglutaminase 5 as a regulator of polyglutamine-expanded ataxin-1
- PMID: 35499073
- PMCID: PMC9057624
- DOI: 10.1172/JCI156616
Cross-species genetic screens identify transglutaminase 5 as a regulator of polyglutamine-expanded ataxin-1
Abstract
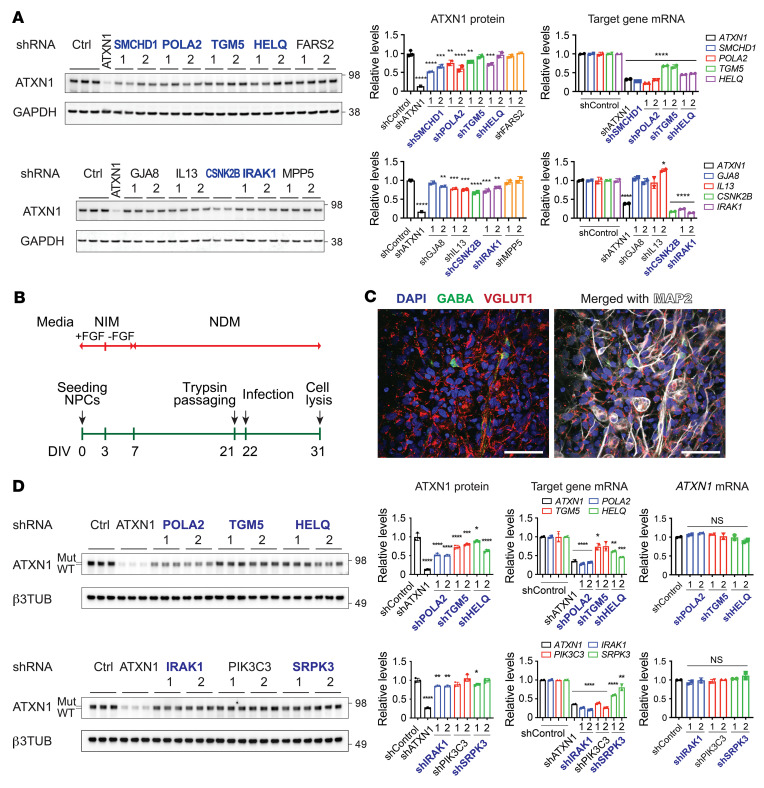
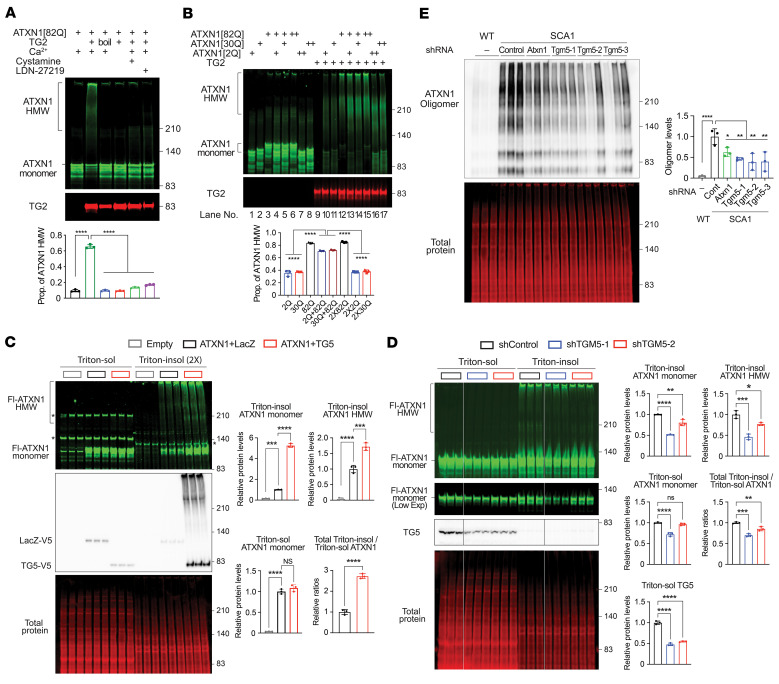
Many neurodegenerative disorders are caused by abnormal accumulation of misfolded proteins. In spinocerebellar ataxia type 1 (SCA1), accumulation of polyglutamine-expanded (polyQ-expanded) ataxin-1 (ATXN1) causes neuronal toxicity. Lowering total ATXN1, especially the polyQ-expanded form, alleviates disease phenotypes in mice, but the molecular mechanism by which the mutant ATXN1 is specifically modulated is not understood. Here, we identified 22 mutant ATXN1 regulators by performing a cross-species screen of 7787 and 2144 genes in human cells and Drosophila eyes, respectively. Among them, transglutaminase 5 (TG5) preferentially regulated mutant ATXN1 over the WT protein. TG enzymes catalyzed cross-linking of ATXN1 in a polyQ-length-dependent manner, thereby preferentially modulating mutant ATXN1 stability and oligomerization. Perturbing Tg in Drosophila SCA1 models modulated mutant ATXN1 toxicity. Moreover, TG5 was enriched in the nuclei of SCA1-affected neurons and colocalized with nuclear ATXN1 inclusions in brain tissue from patients with SCA1. Our work provides a molecular insight into SCA1 pathogenesis and an opportunity for allele-specific targeting for neurodegenerative disorders.
Keywords: Genetic diseases; Genetics; Molecular pathology; Neurodegeneration; Neuroscience.
Conflict of interest statement
Figures





References
-
- Farr SA, et al. Central and peripheral administration of antisense oligonucleotide targeting amyloid-β protein precursor improves learning and memory and reduces neuroinflammatory cytokines in Tg2576 (AβPPswe) mice. J Alzheimers Dis. 2014;40(4):1005–1016. doi: 10.3233/JAD-131883. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
Research Materials
Miscellaneous
