

Assessing in vivo mutation frequencies and creating a high-resolution genome-wide map of fitness costs of Hepatitis C virus
- PMID: 35500034
- PMCID: PMC9113599
- DOI: 10.1371/journal.pgen.1010179
Assessing in vivo mutation frequencies and creating a high-resolution genome-wide map of fitness costs of Hepatitis C virus
Abstract
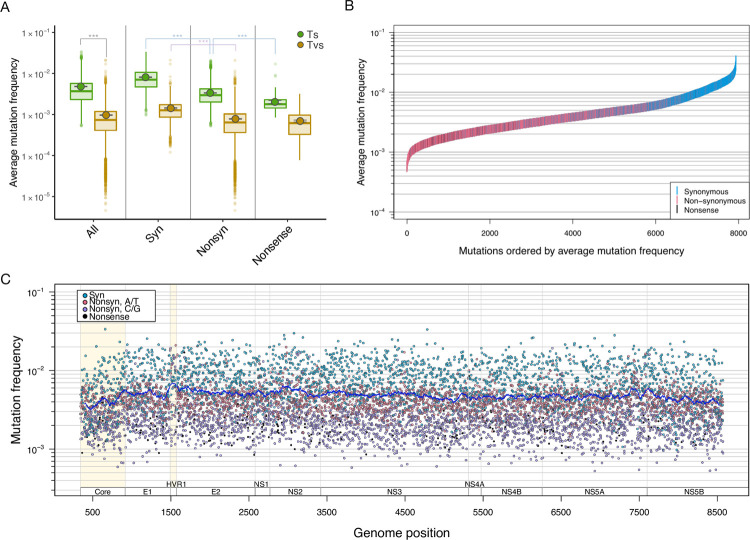
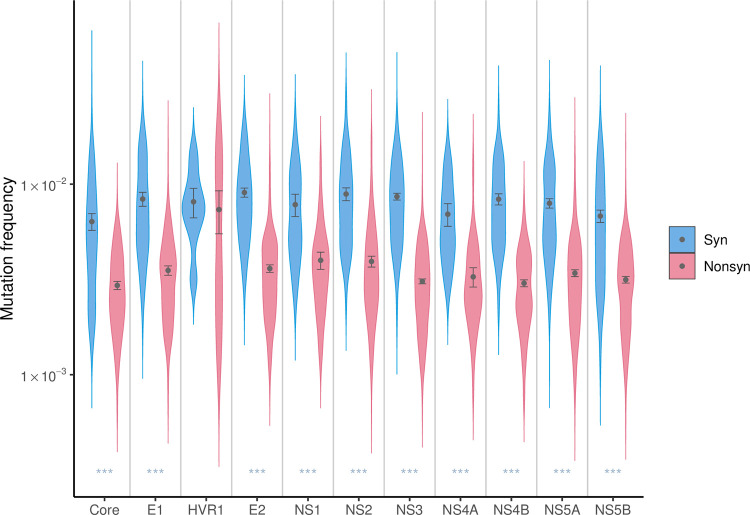
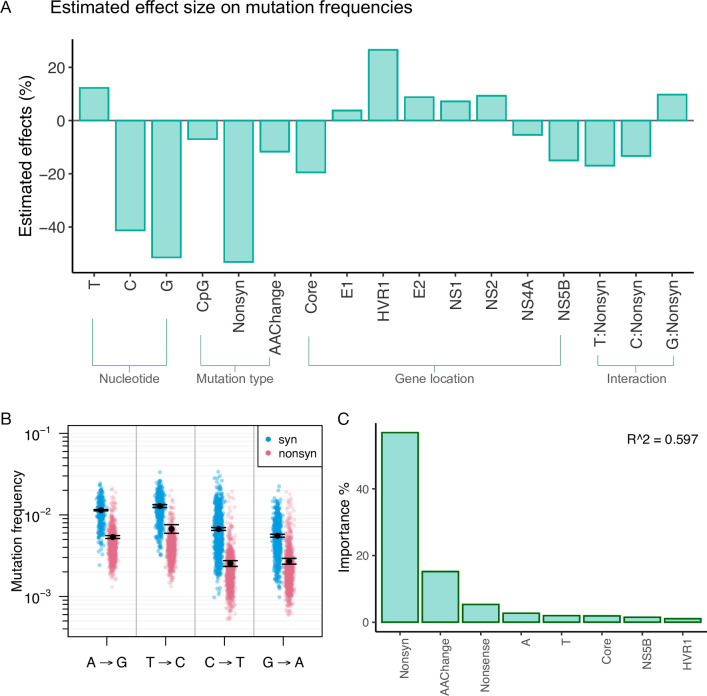
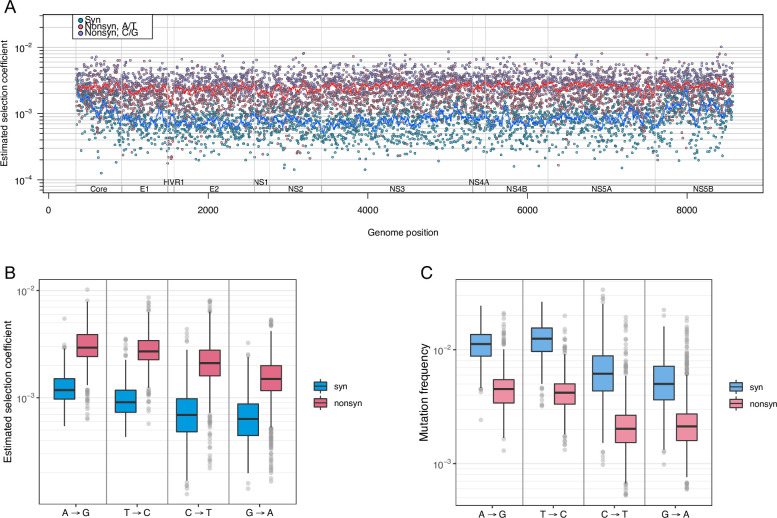
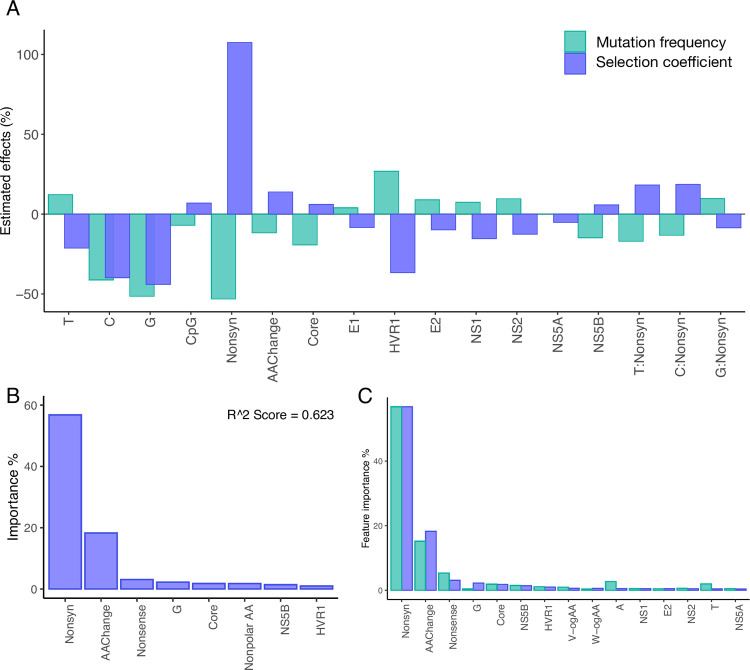
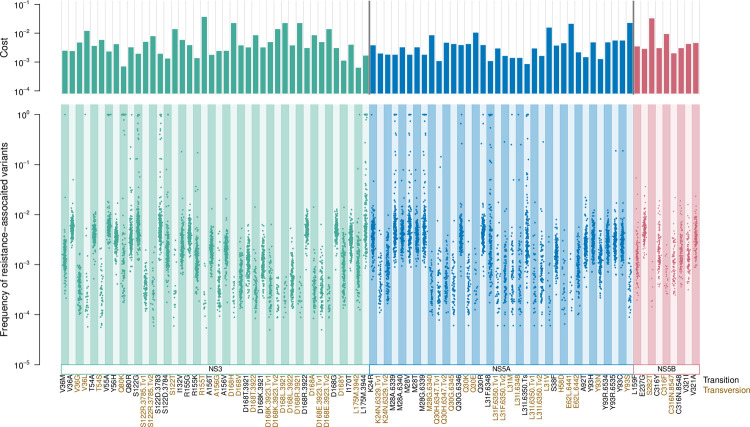
Like many viruses, Hepatitis C Virus (HCV) has a high mutation rate, which helps the virus adapt quickly, but mutations come with fitness costs. Fitness costs can be studied by different approaches, such as experimental or frequency-based approaches. The frequency-based approach is particularly useful to estimate in vivo fitness costs, but this approach works best with deep sequencing data from many hosts are. In this study, we applied the frequency-based approach to a large dataset of 195 patients and estimated the fitness costs of mutations at 7957 sites along the HCV genome. We used beta regression and random forest models to better understand how different factors influenced fitness costs. Our results revealed that costs of nonsynonymous mutations were three times higher than those of synonymous mutations, and mutations at nucleotides A or T had higher costs than those at C or G. Genome location had a modest effect, with lower costs for mutations in HVR1 and higher costs for mutations in Core and NS5B. Resistance mutations were, on average, costlier than other mutations. Our results show that in vivo fitness costs of mutations can be site and virus specific, reinforcing the utility of constructing in vivo fitness cost maps of viral genomes.
Conflict of interest statement
The authors have declared that no competing interests exist.
Figures
References
-
- Bush RM, Bender CA, Subbarao K, Cox NJ, Fitch WM. Predicting the Evolution of Human Influenza A. Science. 1999;286: 1921–1925. Available: https://www.science.org/doi/10.1126/science.286.5446.1921 - DOI - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Medical
