

Exploration of chemical space with partial labeled noisy student self-training and self-supervised graph embedding
- PMID: 35501680
- PMCID: PMC9063120
- DOI: 10.1186/s12859-022-04681-3
Exploration of chemical space with partial labeled noisy student self-training and self-supervised graph embedding
Abstract
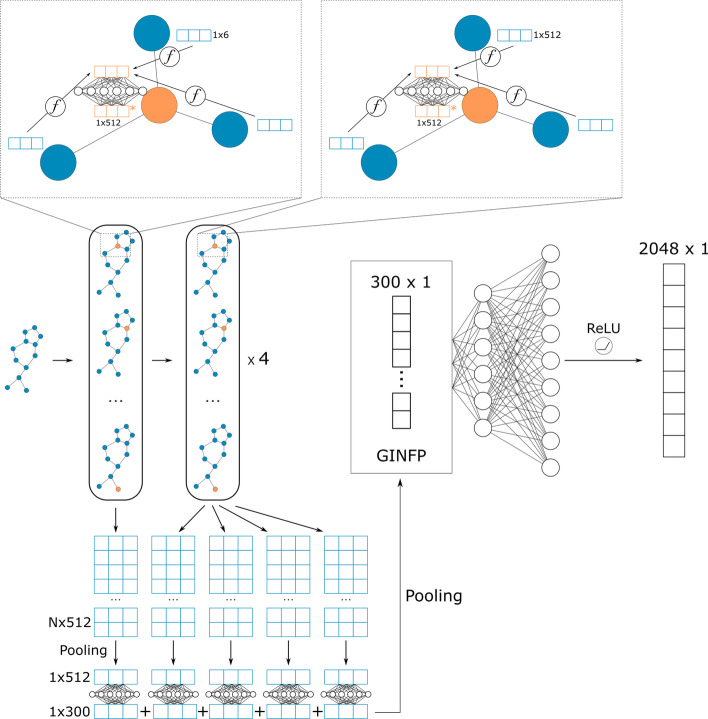
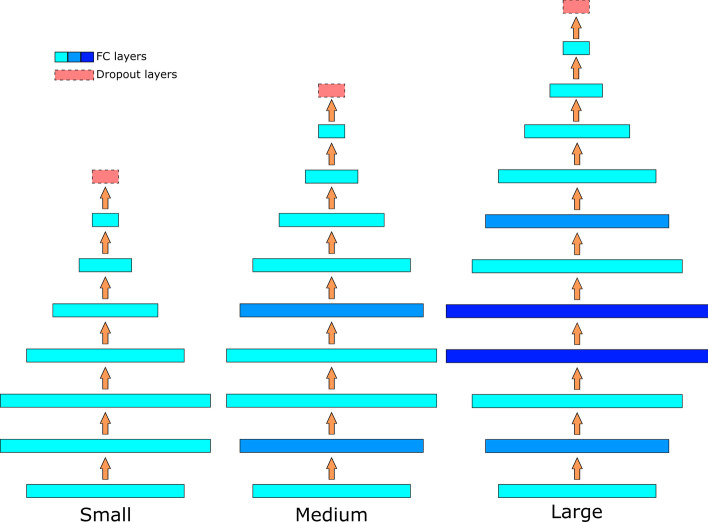
Background: Drug discovery is time-consuming and costly. Machine learning, especially deep learning, shows great potential in quantitative structure-activity relationship (QSAR) modeling to accelerate drug discovery process and reduce its cost. A big challenge in developing robust and generalizable deep learning models for QSAR is the lack of a large amount of data with high-quality and balanced labels. To address this challenge, we developed a self-training method, Partially LAbeled Noisy Student (PLANS), and a novel self-supervised graph embedding, Graph-Isomorphism-Network Fingerprint (GINFP), for chemical compounds representations with substructure information using unlabeled data. The representations can be used for predicting chemical properties such as binding affinity, toxicity, and others. PLANS-GINFP allows us to exploit millions of unlabeled chemical compounds as well as labeled and partially labeled pharmacological data to improve the generalizability of neural network models.
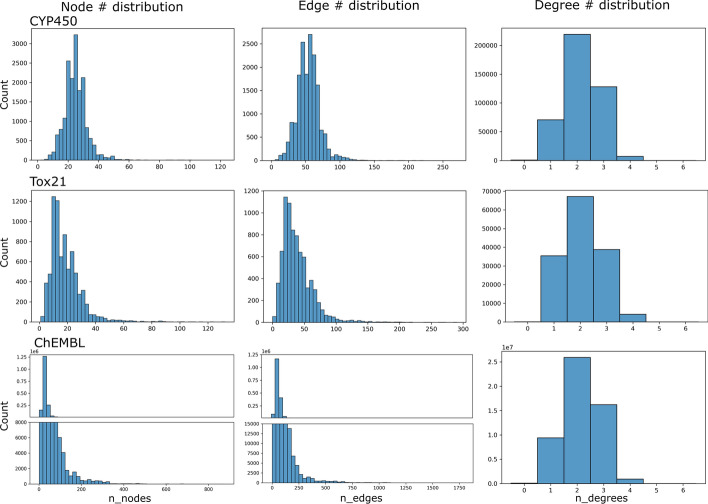
Results: We evaluated the performance of PLANS-GINFP for predicting Cytochrome P450 (CYP450) binding activity in a CYP450 dataset and chemical toxicity in the Tox21 dataset. The extensive benchmark studies demonstrated that PLANS-GINFP could significantly improve the performance in both cases by a large margin. Both PLANS-based self-training and GINFP-based self-supervised learning contribute to the performance improvement.
Conclusion: To better exploit chemical structures as an input for machine learning algorithms, we proposed a self-supervised graph neural network-based embedding method that can encode substructure information. Furthermore, we developed a model agnostic self-training method, PLANS, that can be applied to any deep learning architectures to improve prediction accuracies. PLANS provided a way to better utilize partially labeled and unlabeled data. Comprehensive benchmark studies demonstrated their potentials in predicting drug metabolism and toxicity profiles using sparse, noisy, and imbalanced data. PLANS-GINFP could serve as a general solution to improve the predictive modeling for QSAR modeling.
Keywords: Artificial intelligence; Chemical embedding; Deep neural network; Drug discovery; Drug metabolism; Drug toxicity; Drug-target interaction; Graph neural network; Self-supervised learning; Semi-supervised learning.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures
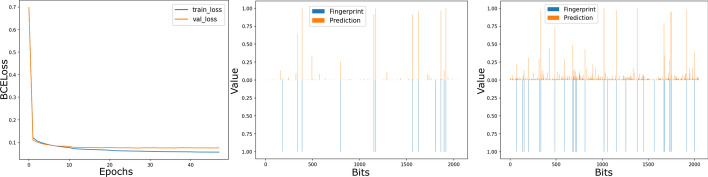
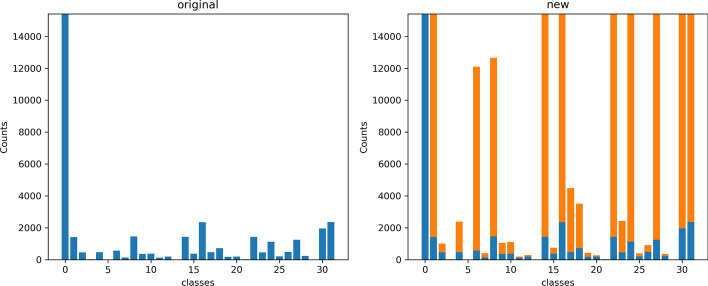
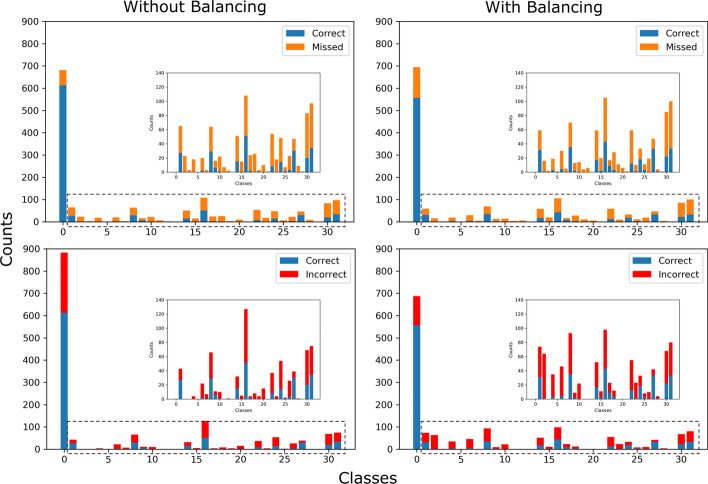
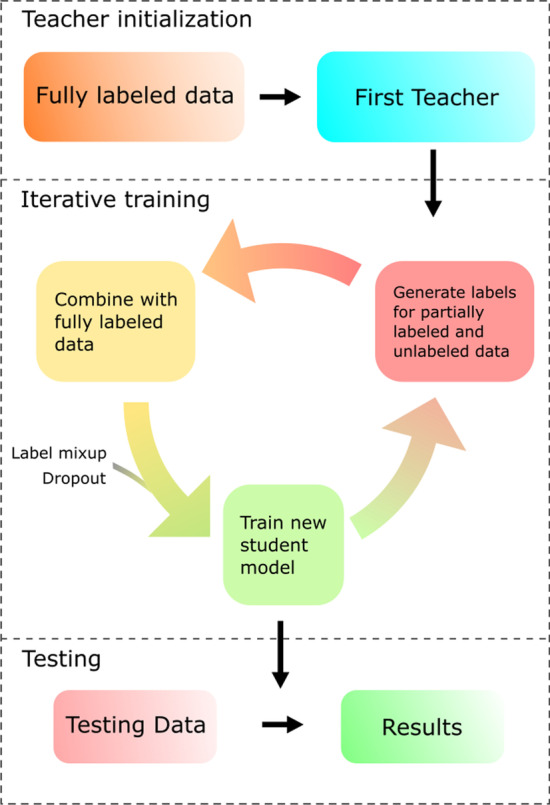
References
-
- Rumelhart DE, McClelland JL, PDP Research Group C, editors. Parallel distributed processing: explorations in the microstructure of cognition, vol 1, foundations. Cambridge: MIT Press; 1986.
-
- Kipf TN, Welling M. Semi-supervised classification with graph convolutional networks. 2016.
-
- Kingma DP, Welling M. Auto-encoding variational bayes.
-
- Kipf TN, Welling M. Variational graph auto-encoders. 2016.
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
