

Parvalbumin neuroplasticity compensates for somatostatin impairment, maintaining cognitive function in Alzheimer's disease
- PMID: 35501886
- PMCID: PMC9063209
- DOI: 10.1186/s40035-022-00300-6
Parvalbumin neuroplasticity compensates for somatostatin impairment, maintaining cognitive function in Alzheimer's disease
Abstract
Background: Patient-to-patient variability in the degree to which β-amyloid, tau and neurodegeneration impact cognitive decline in Alzheimer's disease (AD) complicates disease modeling and treatment. However, the underlying mechanisms leading to cognitive resilience are not resolved. We hypothesize that the variability in cognitive function and loss relates to neuronal resilience of the hippocampal GABAergic network.
Methods: We compared TgF344-AD and non-transgenic littermate rats at 9, 12, and 15 months of age. Neurons, β-amyloid plaques and tau inclusions were quantified in hippocampus and entorhinal cortex. Somatostatin (SST) and parvalbumin (PVB) interneurons were traced to examine hippocampal neuroplasticity and cognition was tested in the Barnes maze.
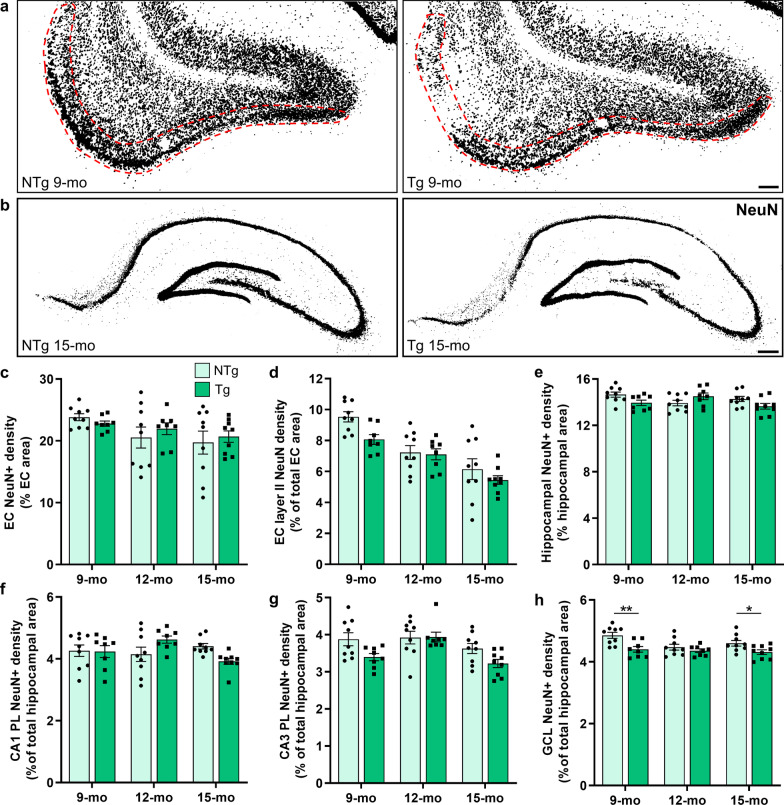
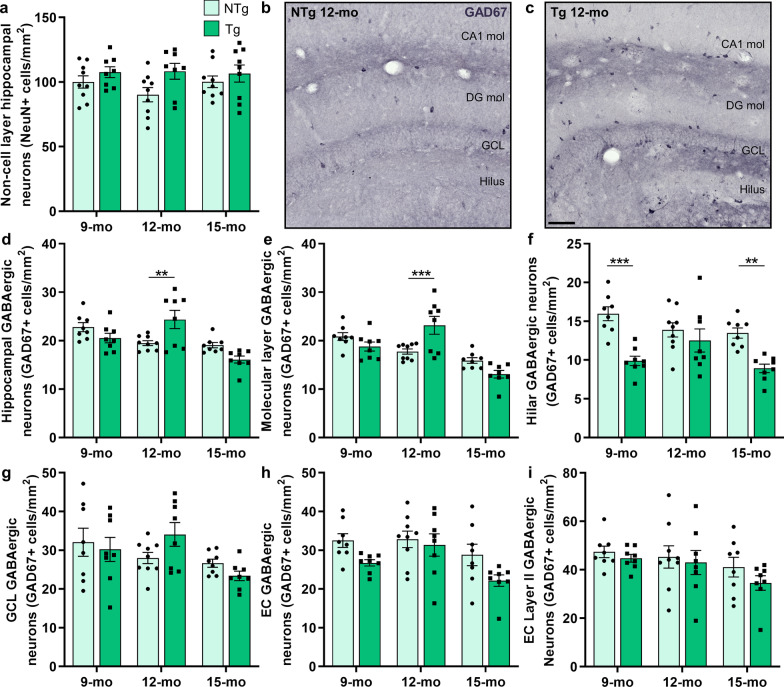
Results: The 9-month-old TgF344-AD rats exhibited loss of neurons in the entorhinal cortex and hippocampus. Hippocampal neuronal compensation was observed in 12-month TgF344-AD rats, with upregulation of GABAergic interneuronal marker. By 15 months, the TgF344-AD rats had robust loss of excitatory and inhibitory neurons. β-Amyloid and tau pathology accumulated continuously across age. SST interneurons exhibited tau inclusions and atrophy from 9 months, whereas PVB interneurons were resilient until 15 months. The hippocampal PVB circuit underwent neuroplastic reorganization with increased dendritic length and complexity in 9- and 12-month-old TgF344-AD rats, before atrophy at 15 months. Strikingly, 12-month-old TgF344-AD rats were resilient in executive function and cognitive flexibility. Cognitive resilience in TgF344-AD rats occurred as maintenance of function between 9 and 12 months of age despite progressive spatial memory deficits, and was sustained by PVB neuroplasticity.
Conclusions: Our results demonstrate the inherent neuronal processes leading to cognitive maintenance, and describe a novel finding of endogenous cognitive resilience in an AD model.
Keywords: Alzheimer’s disease; Barnes maze; Cognitive resilience; GABAergic interneuron; Hippocampus; Neuronal compensation; Parvalbumin; Somatostatin; TgF344-AD rat.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures






Similar articles
-
Quantification and correlation of amyloid-β plaque load, glial activation, GABAergic interneuron numbers, and cognitive decline in the young TgF344-AD rat model of Alzheimer's disease.Front Aging Neurosci. 2025 Feb 12;17:1542229. doi: 10.3389/fnagi.2025.1542229. eCollection 2025. Front Aging Neurosci. 2025. PMID: 40013092 Free PMC article.
-
Regional differences in Alzheimer's disease pathology confound behavioural rescue after amyloid-β attenuation.Brain. 2020 Jan 1;143(1):359-373. doi: 10.1093/brain/awz371. Brain. 2020. PMID: 31782760 Free PMC article.
-
Heightened Hippocampal β-Adrenergic Receptor Function Drives Synaptic Potentiation and Supports Learning and Memory in the TgF344-AD Rat Model during Prodromal Alzheimer's Disease.J Neurosci. 2021 Jun 30;41(26):5747-5761. doi: 10.1523/JNEUROSCI.0119-21.2021. Epub 2021 May 5. J Neurosci. 2021. PMID: 33952633 Free PMC article.
-
Insights From TgF344-AD, a Double Transgenic Rat Model in Alzheimer's Disease Research.Physiol Res. 2025 Mar 21;74(1):1-17. doi: 10.33549/physiolres.935464. Physiol Res. 2025. PMID: 40116546 Free PMC article. Review.
-
α-Melanocyte Stimulating Hormone as a Potential Therapy for Alzheimer`s Disease.Curr Alzheimer Res. 2017;14(1):18-29. doi: 10.2174/1567205013666160819130641. Curr Alzheimer Res. 2017. PMID: 27539595 Review.
Cited by
-
Associations of plasma phosphorylated tau181 and neurofilament light chain with brain amyloid burden and cognition in objectively defined subtle cognitive decline patients.CNS Neurosci Ther. 2022 Dec;28(12):2195-2205. doi: 10.1111/cns.13962. Epub 2022 Sep 8. CNS Neurosci Ther. 2022. PMID: 36074638 Free PMC article.
-
High caloric intake improves neuronal metabolism and functional hyperemia in a rat model of early AD pathology.Theranostics. 2024 Oct 28;14(19):7405-7423. doi: 10.7150/thno.98793. eCollection 2024. Theranostics. 2024. PMID: 39659583 Free PMC article.
-
Linking Social Cognition, Parvalbumin Interneurons, and Oxytocin in Alzheimer's Disease: An Update.J Alzheimers Dis. 2023;96(3):861-875. doi: 10.3233/JAD-230333. J Alzheimers Dis. 2023. PMID: 37980658 Free PMC article. Review.
-
Local and long-range GABAergic circuits in hippocampal area CA1 and their link to Alzheimer's disease.Front Neural Circuits. 2023 Sep 29;17:1223891. doi: 10.3389/fncir.2023.1223891. eCollection 2023. Front Neural Circuits. 2023. PMID: 37841892 Free PMC article. Review.
-
Resilience to Alzheimer's disease associates with alterations in perineuronal nets.Alzheimers Dement. 2025 Feb;21(2):e14504. doi: 10.1002/alz.14504. Epub 2024 Dec 31. Alzheimers Dement. 2025. PMID: 39737731 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
