

Acute axon damage and demyelination are mitigated by 4-aminopyridine (4-AP) therapy after experimental traumatic brain injury
- PMID: 35501931
- PMCID: PMC9059462
- DOI: 10.1186/s40478-022-01366-z
Acute axon damage and demyelination are mitigated by 4-aminopyridine (4-AP) therapy after experimental traumatic brain injury
Abstract
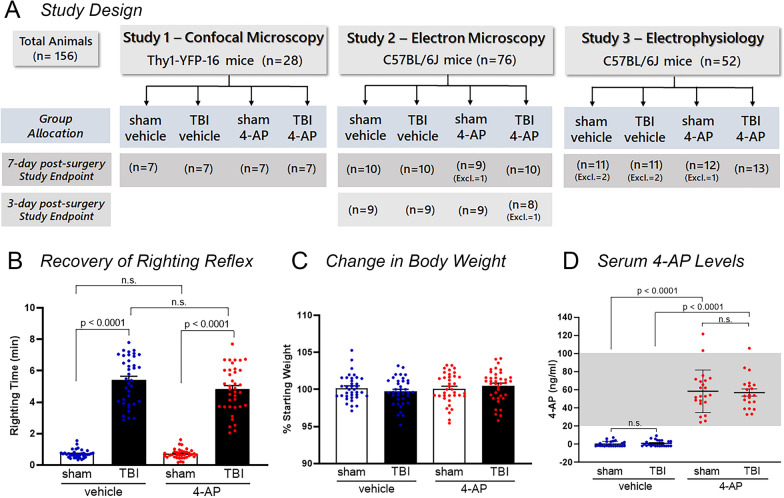
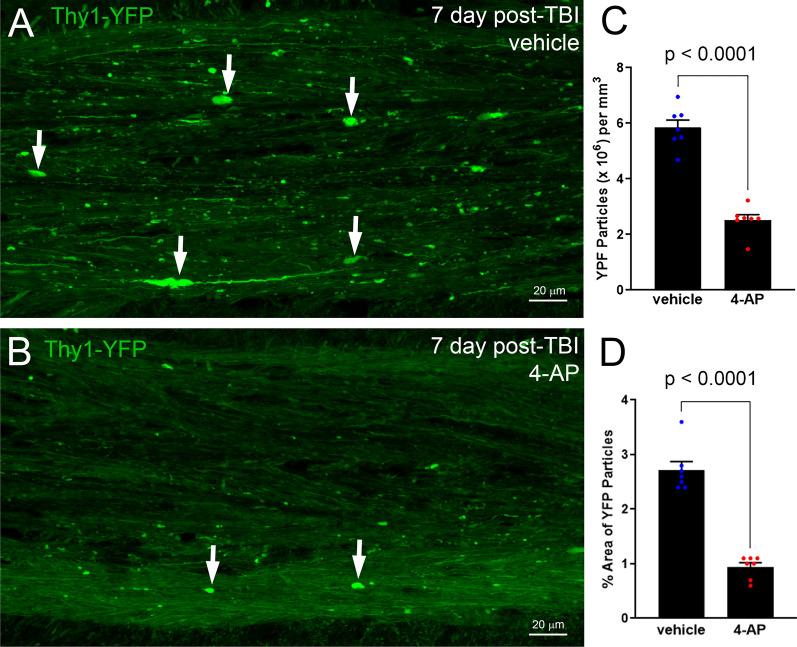
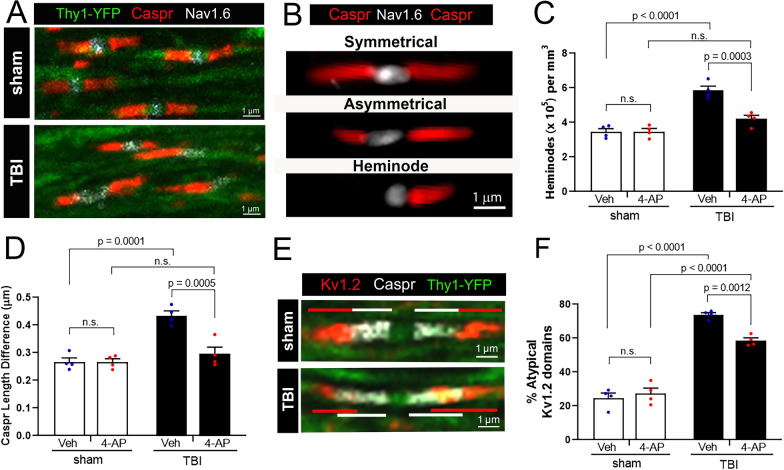
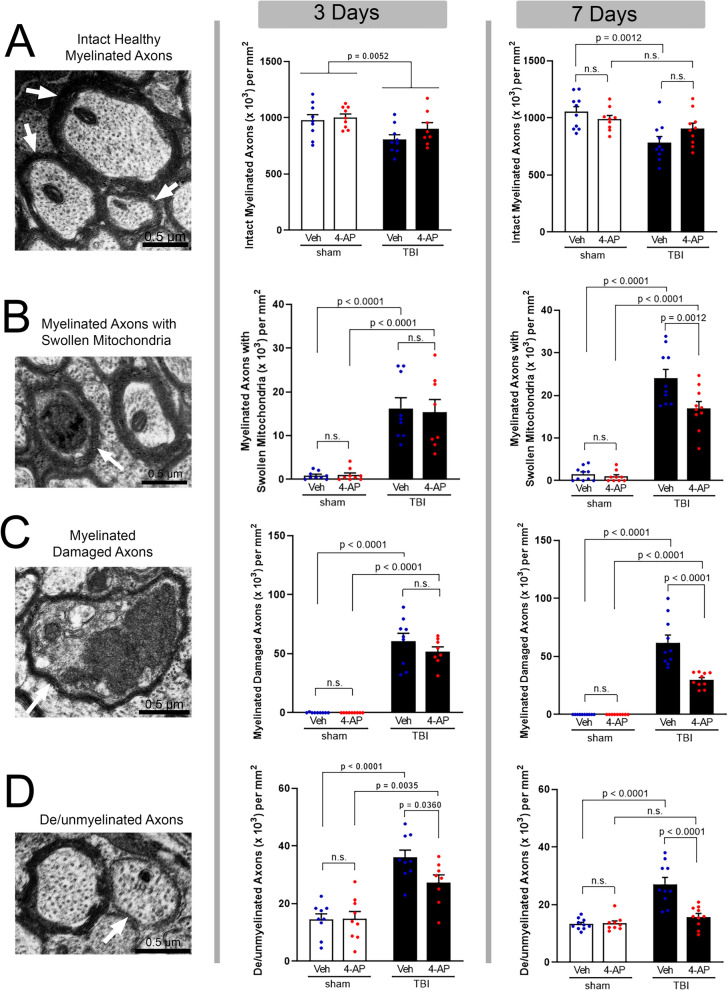
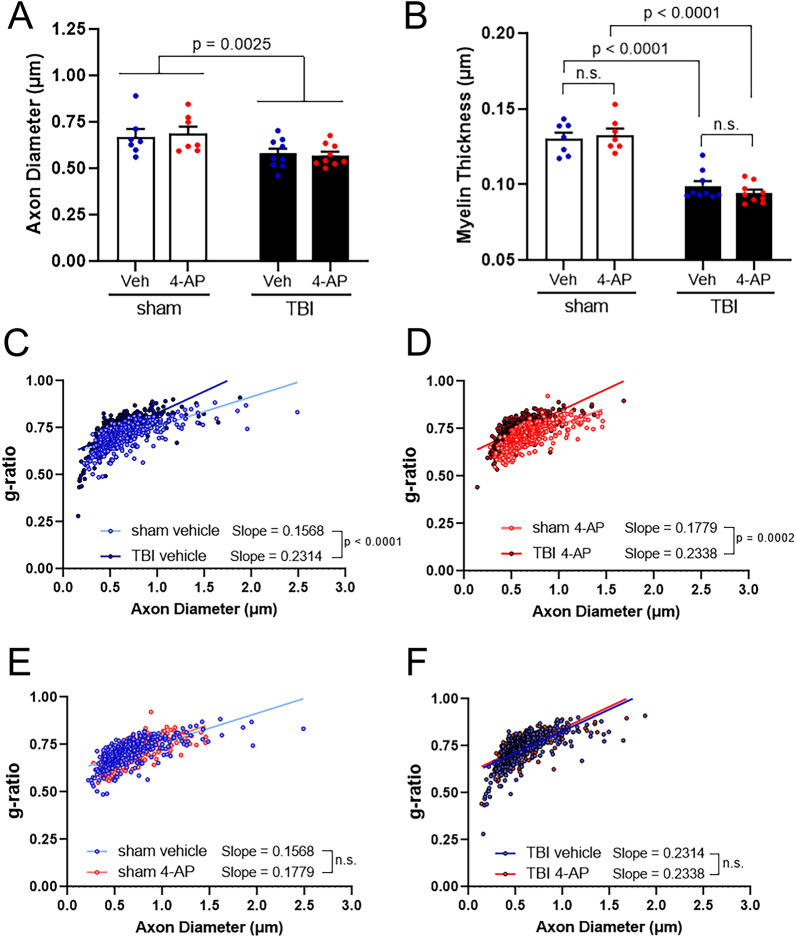
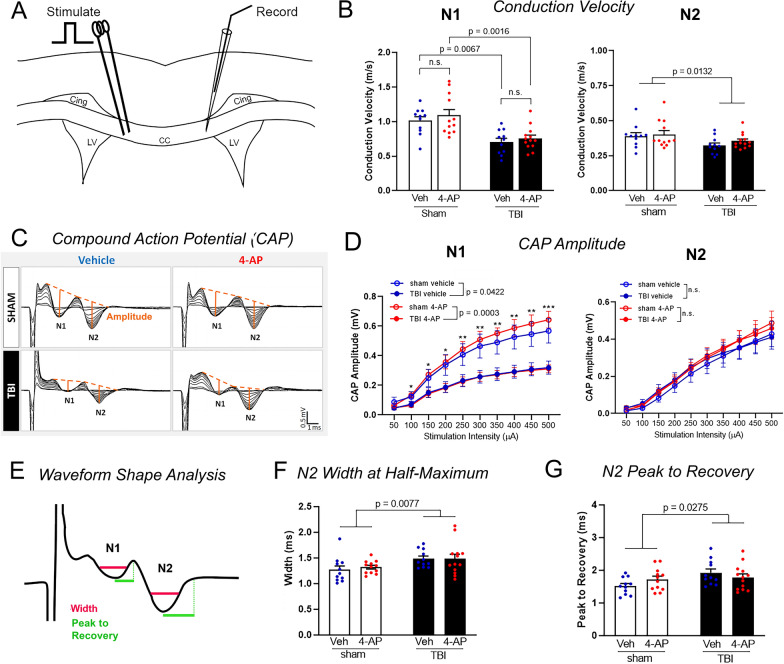
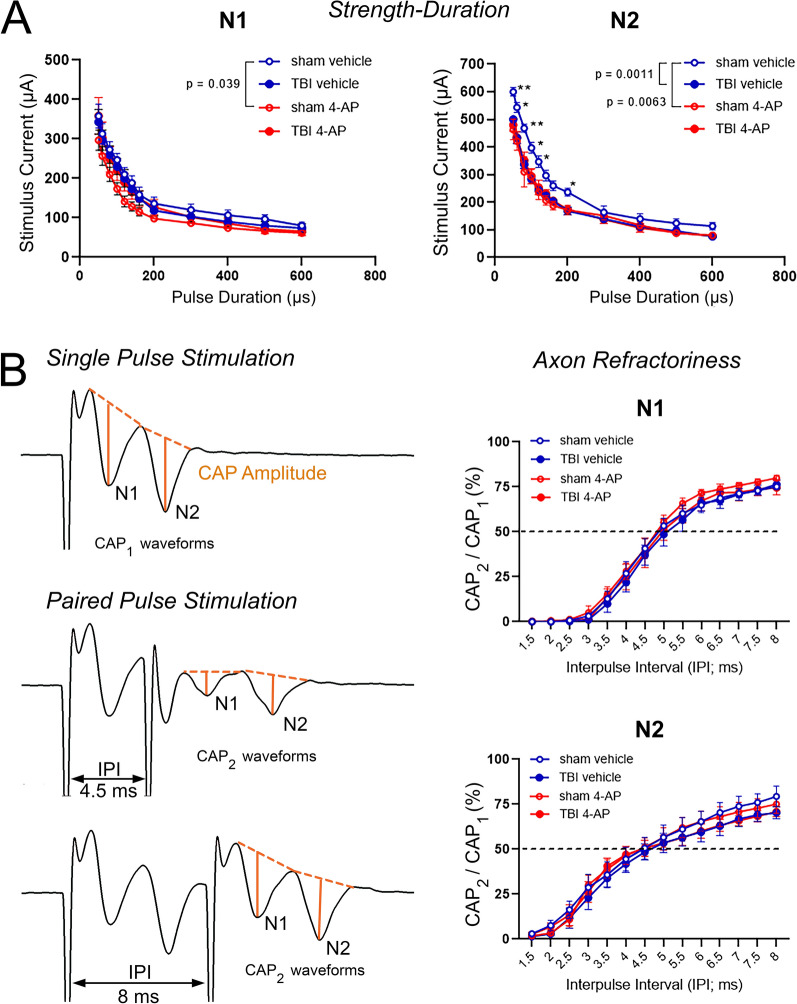
Damage to long axons in white matter tracts is a major pathology in closed head traumatic brain injury (TBI). Acute TBI treatments are needed that protect against axon damage and promote recovery of axon function to prevent long term symptoms and neurodegeneration. Our prior characterization of axon damage and demyelination after TBI led us to examine repurposing of 4-aminopyridine (4-AP), an FDA-approved inhibitor of voltage-gated potassium (Kv) channels. 4-AP is currently indicated to provide symptomatic relief for patients with chronic stage multiple sclerosis, which involves axon damage and demyelination. We tested clinically relevant dosage of 4-AP as an acute treatment for experimental TBI and found multiple benefits in corpus callosum axons. This randomized, controlled pre-clinical study focused on the first week after TBI, when axons are particularly vulnerable. 4-AP treatment initiated one day post-injury dramatically reduced axon damage detected by intra-axonal fluorescence accumulations in Thy1-YFP mice of both sexes. Detailed electron microscopy in C57BL/6 mice showed that 4-AP reduced pathological features of mitochondrial swelling, cytoskeletal disruption, and demyelination at 7 days post-injury. Furthermore, 4-AP improved the molecular organization of axon nodal regions by restoring disrupted paranode domains and reducing Kv1.2 channel dispersion. 4-AP treatment did not resolve deficits in action potential conduction across the corpus callosum, based on ex vivo electrophysiological recordings at 7 days post-TBI. Thus, this first study of 4-AP effects on axon damage in the acute period demonstrates a significant decrease in multiple pathological hallmarks of axon damage after experimental TBI.
Keywords: 4-aminopyridine; Demyelination; Electrophysiology; Kv1.2; Traumatic brain injury; White matter.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing financial interests.
Figures
References
-
- Andravizou A, Dardiotis E, Artemiadis A, Sokratous M, Siokas V, Tsouris Z, Aloizou AM, Nikolaidis I, Bakirtzis C, Tsivgoulis G, et al. Brain atrophy in multiple sclerosis: mechanisms, clinical relevance and treatment options. Auto Immun Highlights. 2019;10:7. doi: 10.1186/s13317-019-0117-5. - DOI - PMC - PubMed
-
- Balan IS, Saladino AJ, Aarabi B, Castellani RJ, Wade C, Stein DM, Eisenberg HM, Chen HH, Fiskum G. Cellular alterations in human traumatic brain injury: changes in mitochondrial morphology reflect regional levels of injury severity. J Neurotrauma. 2013;30:367–381. doi: 10.1089/neu.2012.2339. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Other Literature Sources
Medical
Miscellaneous
