

The effects of age, sex, weight, and breed on canid methylomes
- PMID: 35502722
- PMCID: PMC9586589
- DOI: 10.1080/15592294.2022.2069385
The effects of age, sex, weight, and breed on canid methylomes
Abstract
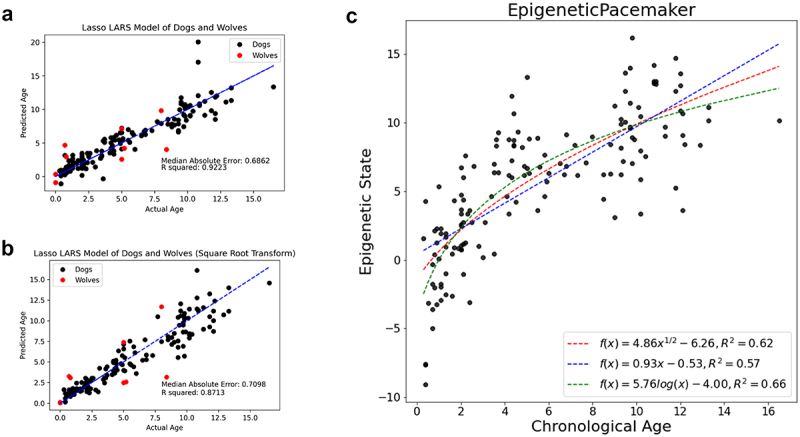
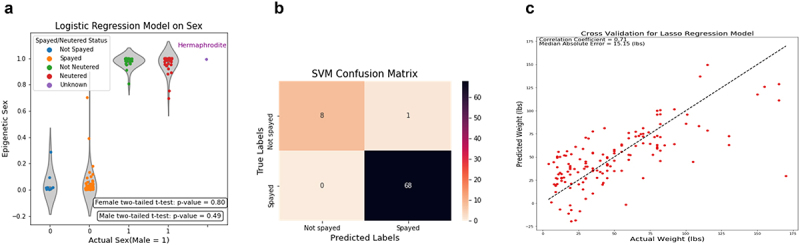
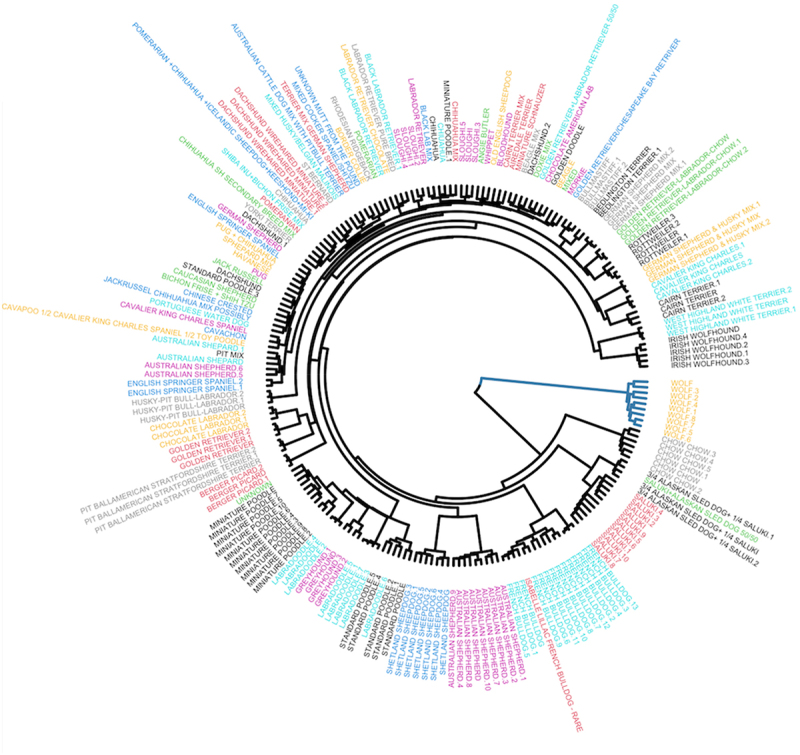
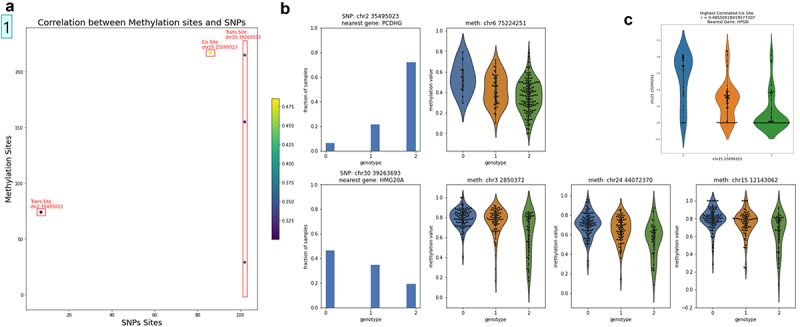
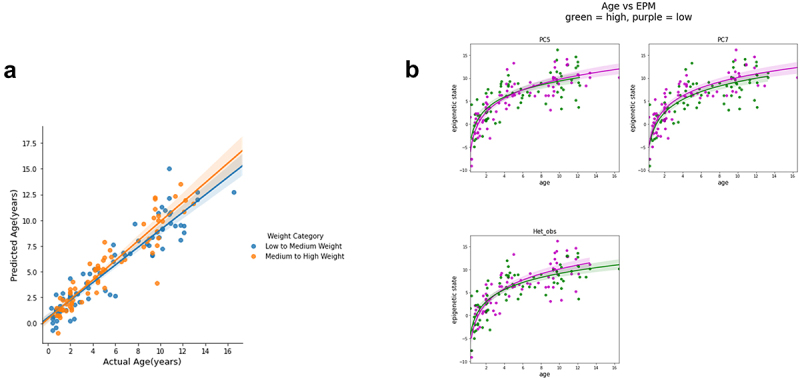
Unlike genomes, which are static throughout the lifespan of an organism, DNA methylomes are dynamic. To study these dynamics, we developed quantitative models that measure the effect of multiple factors on DNA methylomes including, age, sex, weight, and genetics. We conducted our study in canids, which prove to be an ideal species to assess epigenetic moderators due to their extreme variability in size and well-characterized genetic structure. We collected buccal swabs from 217 canids (207 domestic dogs and 10 grey wolves) and used targeted bisulphite sequencing to measure methylomes. We also measured genotypes at over one thousand single nucleotide polymorphisms (SNPs). As expected, we found that DNA methylomes are strongly associated with age, enabling the construction of epigenetic clocks. However, we also identify novel associations between methylomes and sex, weight, and sterilization status, leading to accurate models that predict these factors. Methylomes are also affected by genetics, and we observe multiple associations between SNP loci and methylated CpGs. Finally, we show that several factors moderate the relationship between epigenetic ages and real ages, such as body weight, which increases epigenetic ageing. In conclusion, we demonstrate that the plasticity of DNA methylomes is impacted by myriad genetics and physiological factors, and that DNA methylation biomarkers are accurate predictors of age, sex and sterilization status.
Keywords: DNA methylation; canids; dogs; epigenetic clock.
Conflict of interest statement
No potential conflict of interest was reported by the author(s).
Figures
Similar articles
-
DNA methylation clocks for dogs and humans.Proc Natl Acad Sci U S A. 2022 May 24;119(21):e2120887119. doi: 10.1073/pnas.2120887119. Epub 2022 May 17. Proc Natl Acad Sci U S A. 2022. PMID: 35580182 Free PMC article.
-
An epigenetic aging clock for dogs and wolves.Aging (Albany NY). 2017 Mar 28;9(3):1055-1068. doi: 10.18632/aging.101211. Aging (Albany NY). 2017. PMID: 28373601 Free PMC article.
-
Whole-Genome Bisulfite Sequencing and Epigenetic Variation in Cereal Methylomes.Methods Mol Biol. 2020;2072:119-128. doi: 10.1007/978-1-4939-9865-4_10. Methods Mol Biol. 2020. PMID: 31541442
-
Epigenetic Clock: Just a Convenient Marker or an Active Driver of Aging?Adv Exp Med Biol. 2019;1178:175-206. doi: 10.1007/978-3-030-25650-0_10. Adv Exp Med Biol. 2019. PMID: 31493228 Review.
-
Making sense of the ageing methylome.Nat Rev Genet. 2022 Oct;23(10):585-605. doi: 10.1038/s41576-022-00477-6. Epub 2022 May 2. Nat Rev Genet. 2022. PMID: 35501397 Review.
Cited by
-
Association of DNA methylation with energy and fear-related behaviors in canines.Front Psychol. 2022 Dec 14;13:1025494. doi: 10.3389/fpsyg.2022.1025494. eCollection 2022. Front Psychol. 2022. PMID: 36591016 Free PMC article.
-
Co-analysis of methylation platforms for signatures of biological aging in the domestic dog reveals previously unexplored confounding factors.Aging (Albany NY). 2024 Jul 9;16(13):10724-10748. doi: 10.18632/aging.206012. Epub 2024 Jul 9. Aging (Albany NY). 2024. PMID: 38985449 Free PMC article.
-
DNA methylation entropy is a biomarker for aging.Aging (Albany NY). 2025 Mar 12;17(3):685-698. doi: 10.18632/aging.206220. Epub 2025 Mar 12. Aging (Albany NY). 2025. PMID: 40096548 Free PMC article.
-
DNA methylation and chromatin accessibility predict age in the domestic dog.Aging Cell. 2024 Apr;23(4):e14079. doi: 10.1111/acel.14079. Epub 2024 Jan 23. Aging Cell. 2024. PMID: 38263575 Free PMC article.
-
Age-correlated changes in the canine oral microbiome.Front Microbiol. 2024 Jul 16;15:1426691. doi: 10.3389/fmicb.2024.1426691. eCollection 2024. Front Microbiol. 2024. PMID: 39081893 Free PMC article.
References
-
- Gilmore KM, Greer KA.. Why is the dog an ideal model for aging research? Exp Gerontol. Aging in the Wild: Insights from Free-Living and Non-Model organisms. 2015;71:14–20. - PubMed
MeSH terms
LinkOut - more resources
Full Text Sources
Other Literature Sources
Research Materials