

BCG therapy downregulates HLA-I on malignant cells to subvert antitumor immune responses in bladder cancer
- PMID: 35503263
- PMCID: PMC9197524
- DOI: 10.1172/JCI145666
BCG therapy downregulates HLA-I on malignant cells to subvert antitumor immune responses in bladder cancer
Abstract
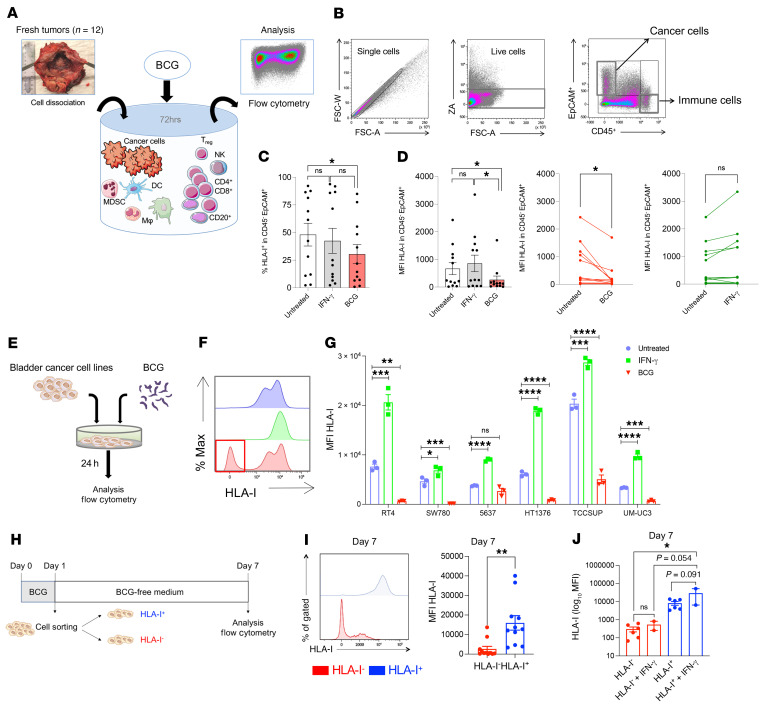
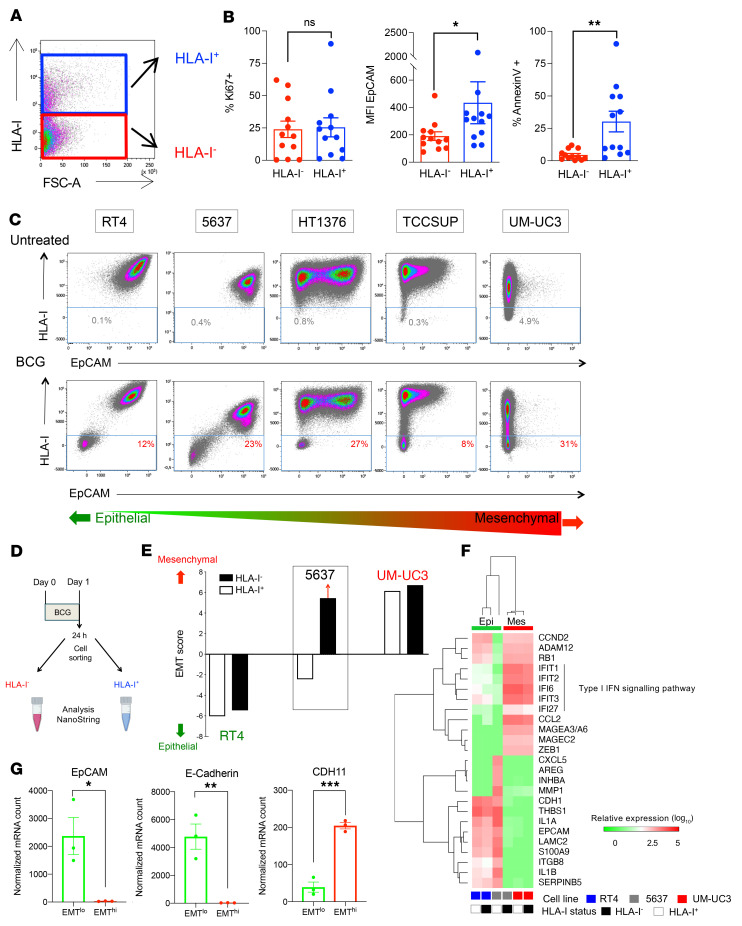
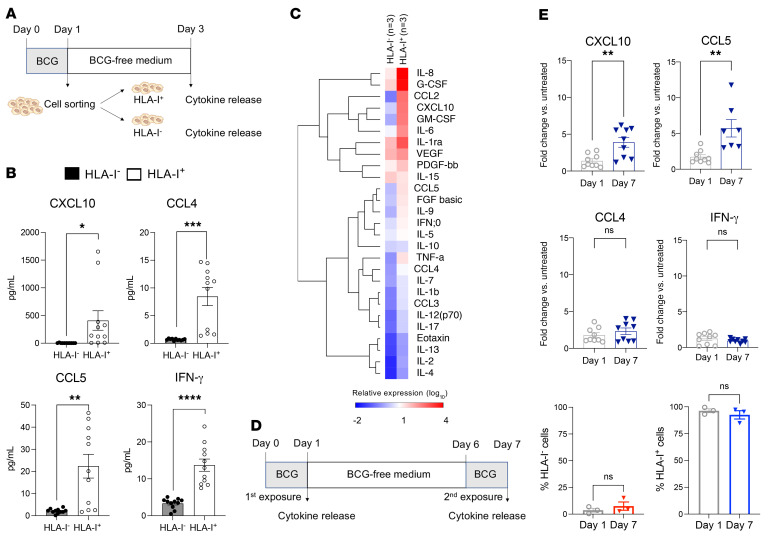
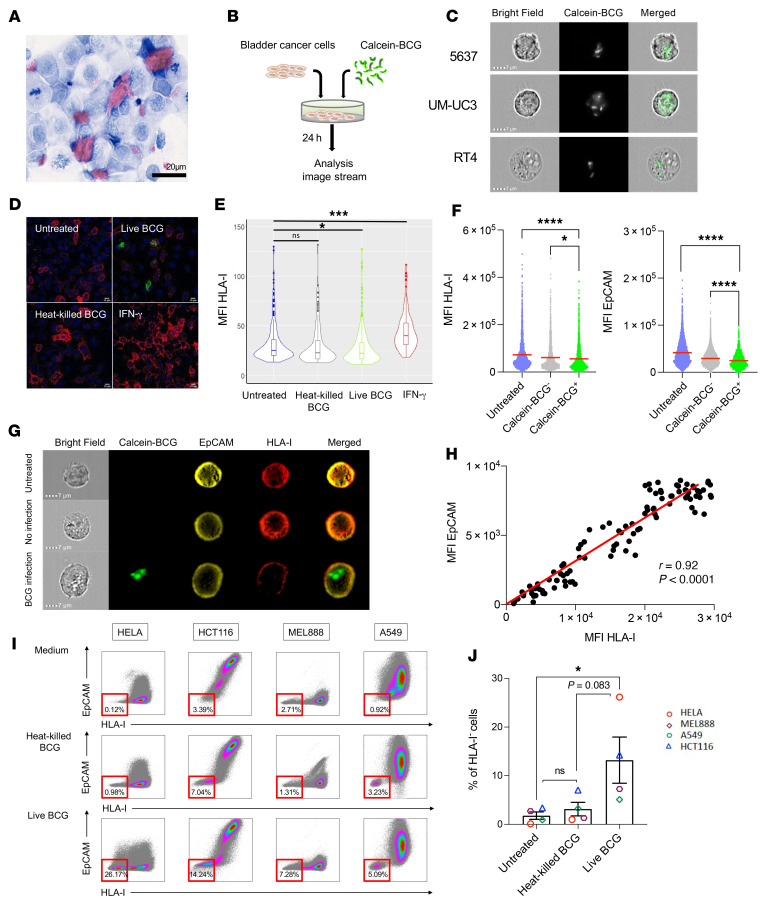
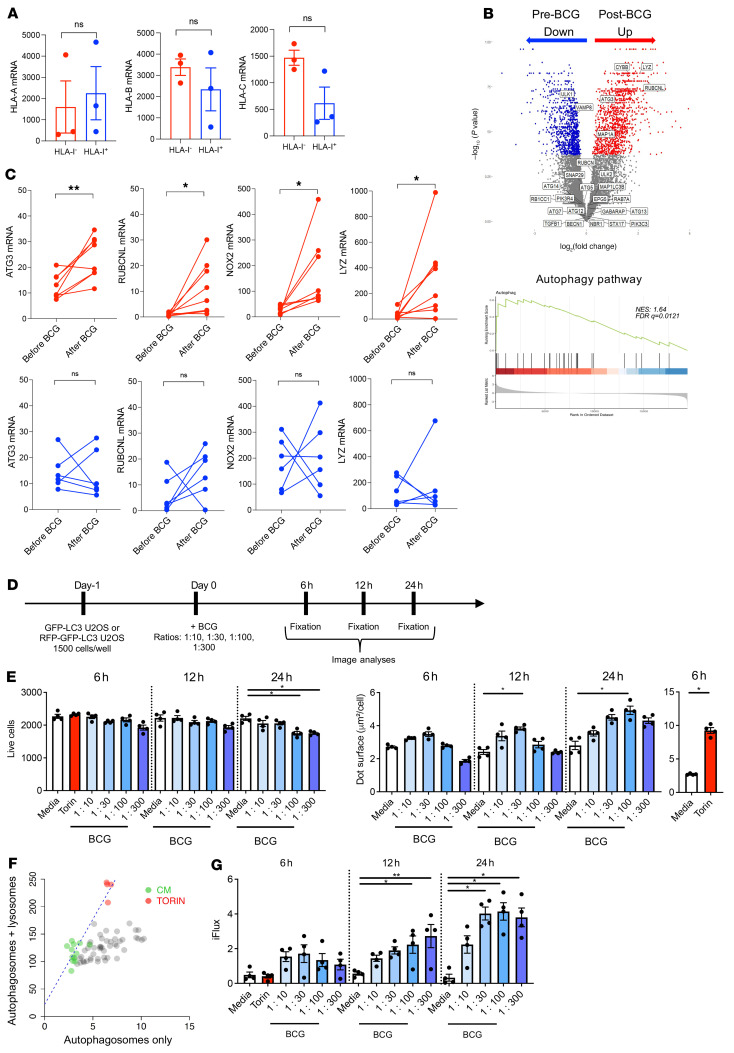
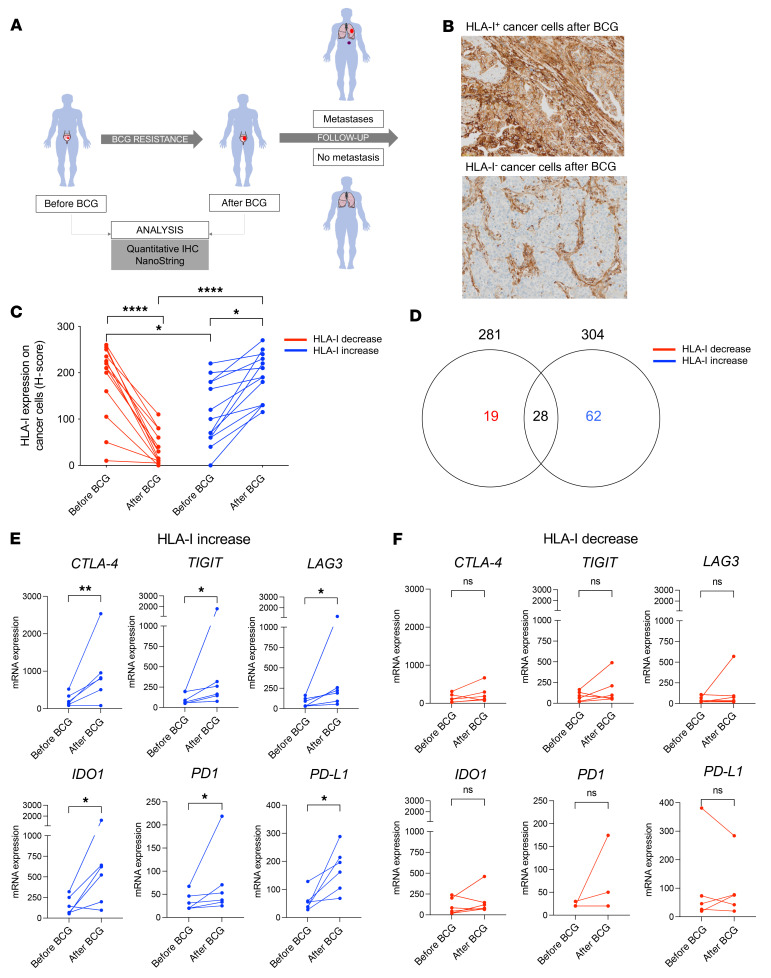
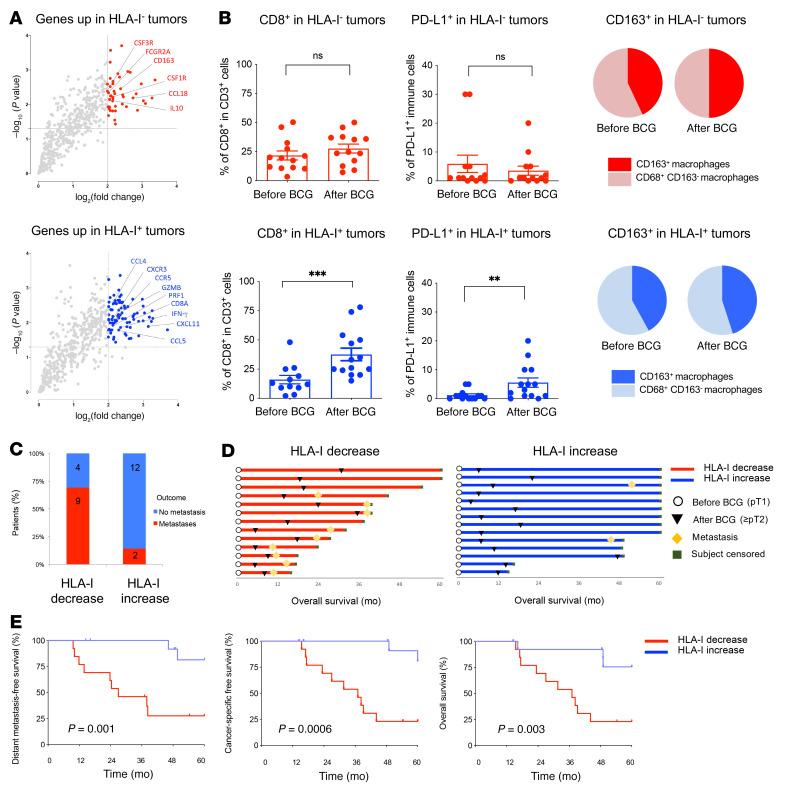
Patients with high-risk, nonmuscle-invasive bladder cancer (NMIBC) frequently relapse after standard intravesical bacillus Calmette-Guérin (BCG) therapy and may have a dismal outcome. The mechanisms of resistance to such immunotherapy remain poorly understood. Here, using cancer cell lines, freshly resected human bladder tumors, and samples from cohorts of patients with bladder cancer before and after BCG therapy, we demonstrate 2 distinct patterns of immune subversion upon BCG relapse. In the first pattern, intracellular BCG infection of cancer cells induced a posttranscriptional downregulation of HLA-I membrane expression via inhibition of autophagy flux. Patients with HLA-I-deficient cancer cells following BCG therapy had a myeloid immunosuppressive tumor microenvironment (TME) with epithelial-mesenchymal transition (EMT) characteristics and dismal outcomes. Conversely, patients with HLA-I-proficient cancer cells after BCG therapy presented with CD8+ T cell tumor infiltrates, upregulation of inflammatory cytokines, and immune checkpoint-inhibitory molecules. The latter patients had a very favorable outcome. We surmise that HLA-I expression in bladder cancers at relapse following BCG does not result from immunoediting but rather from an immune subversion process directly induced by BCG on cancer cells, which predicts a dismal prognosis. HLA-I scoring of cancer cells by IHC staining can be easily implemented by pathologists in routine practice to stratify future treatment strategies for patients with urothelial cancer.
Keywords: Bacterial infections; Cancer; Immunology; MHC class 1; Oncology.
Conflict of interest statement
Figures
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
