

Y-Chromosome Gene, Uty, Protects Against Pulmonary Hypertension by Reducing Proinflammatory Chemokines
- PMID: 35504005
- PMCID: PMC9887415
- DOI: 10.1164/rccm.202110-2309OC
Y-Chromosome Gene, Uty, Protects Against Pulmonary Hypertension by Reducing Proinflammatory Chemokines
Abstract
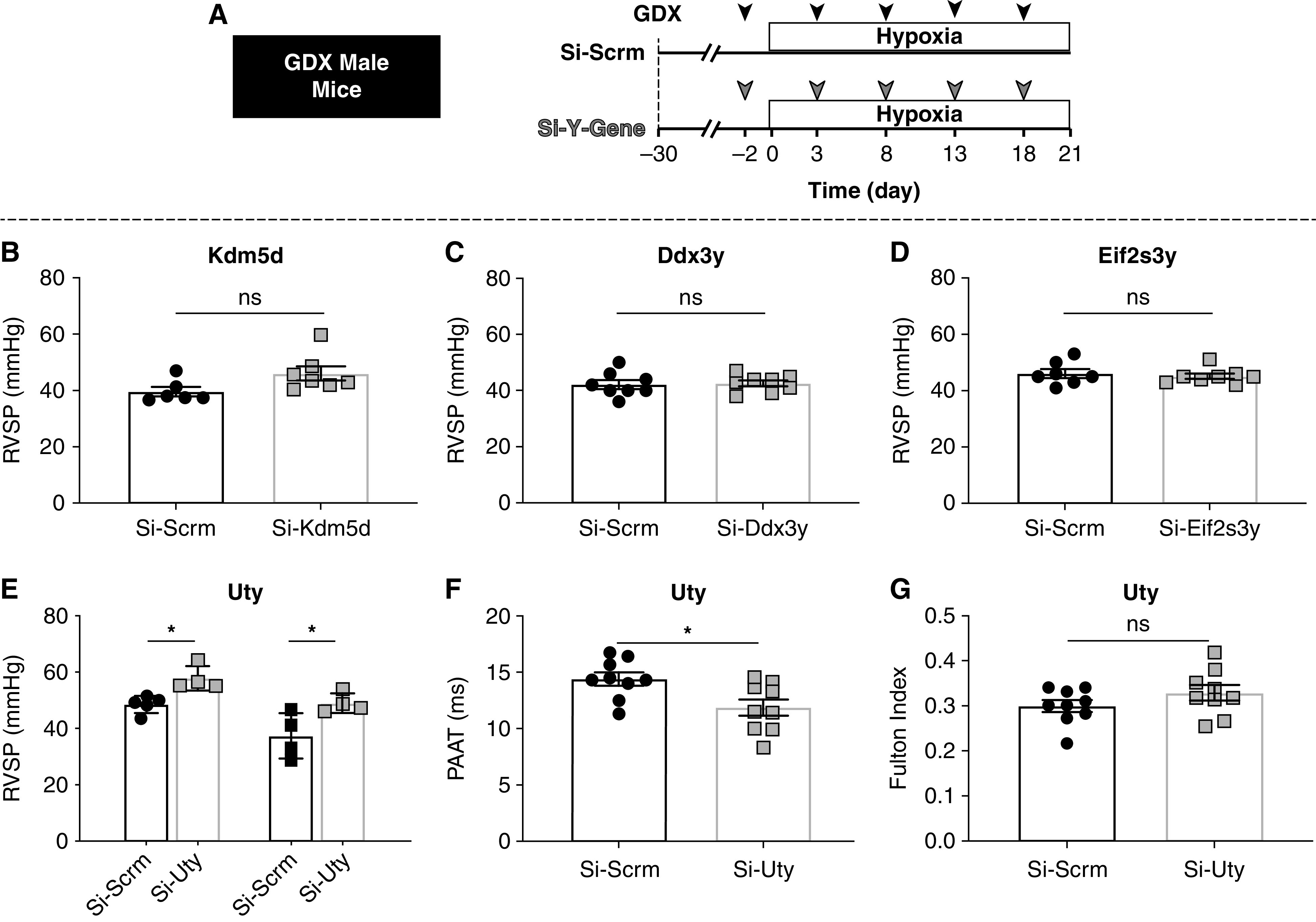
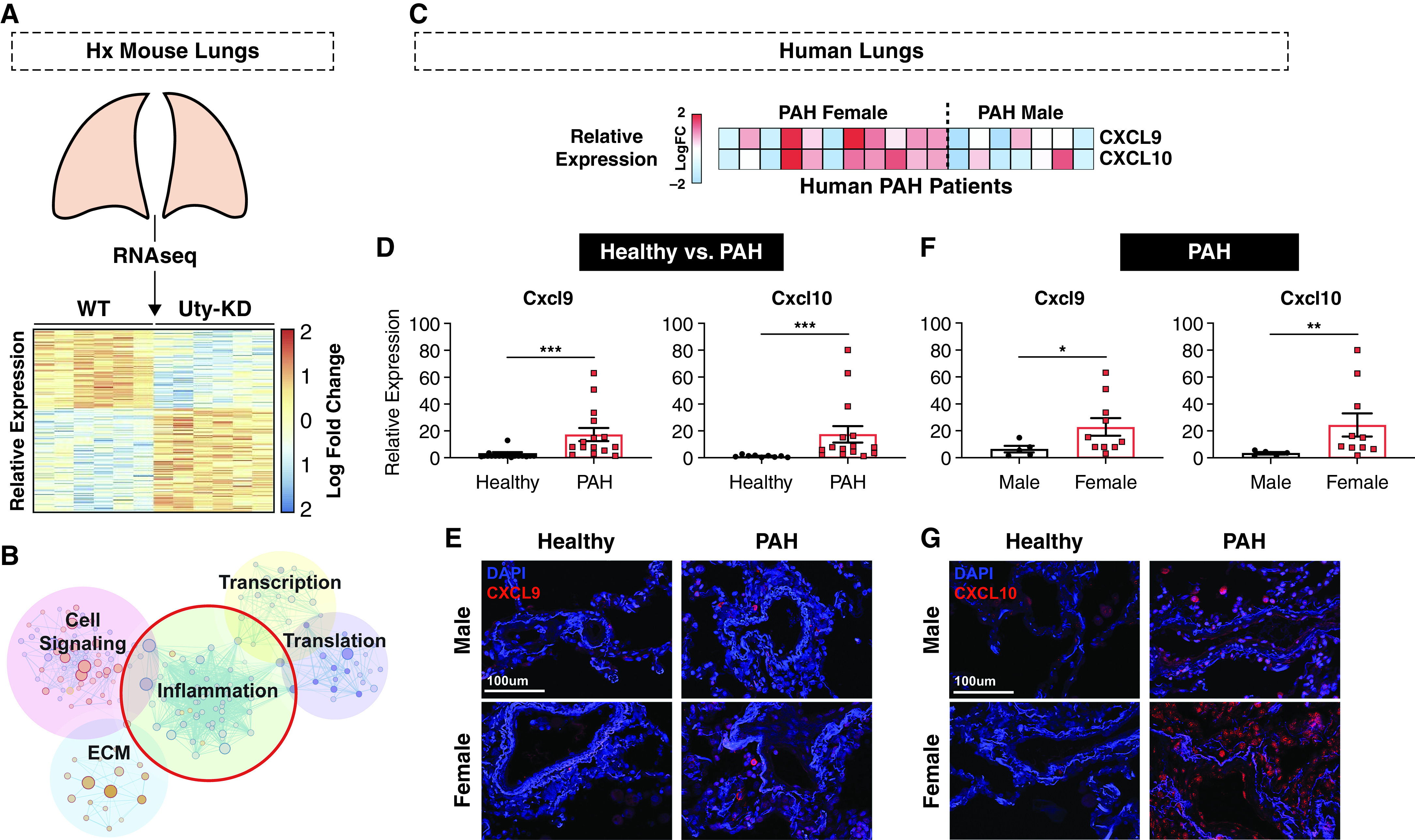
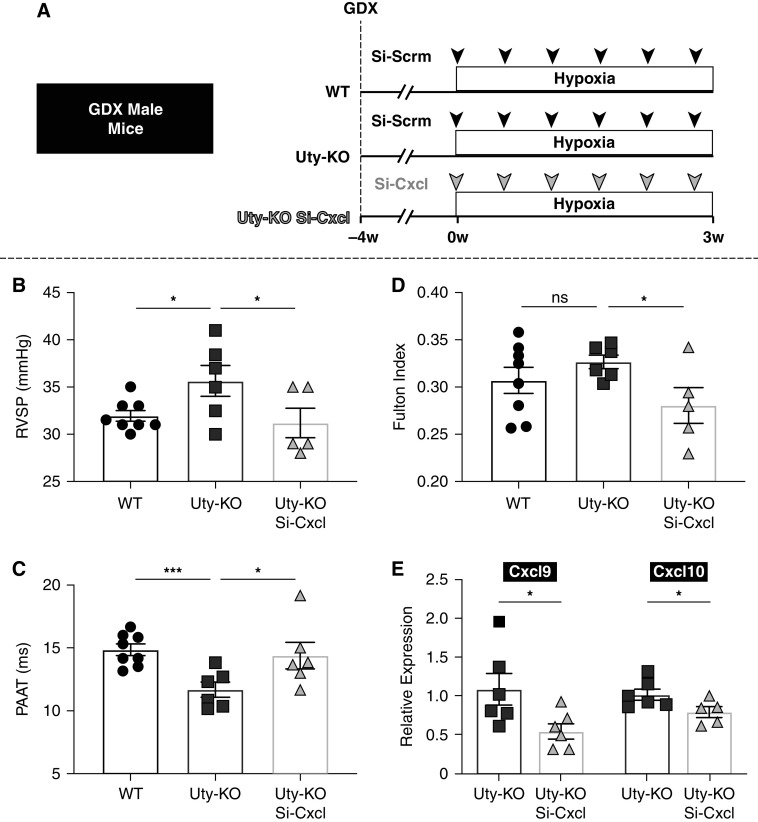
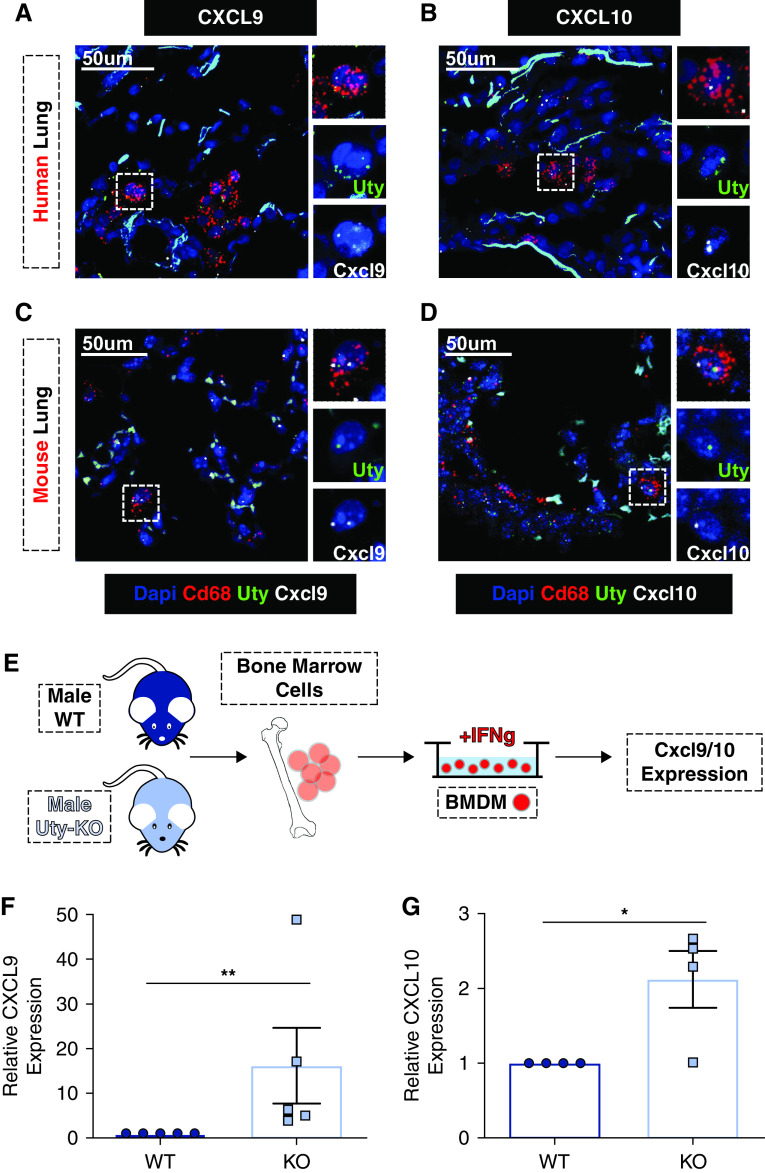
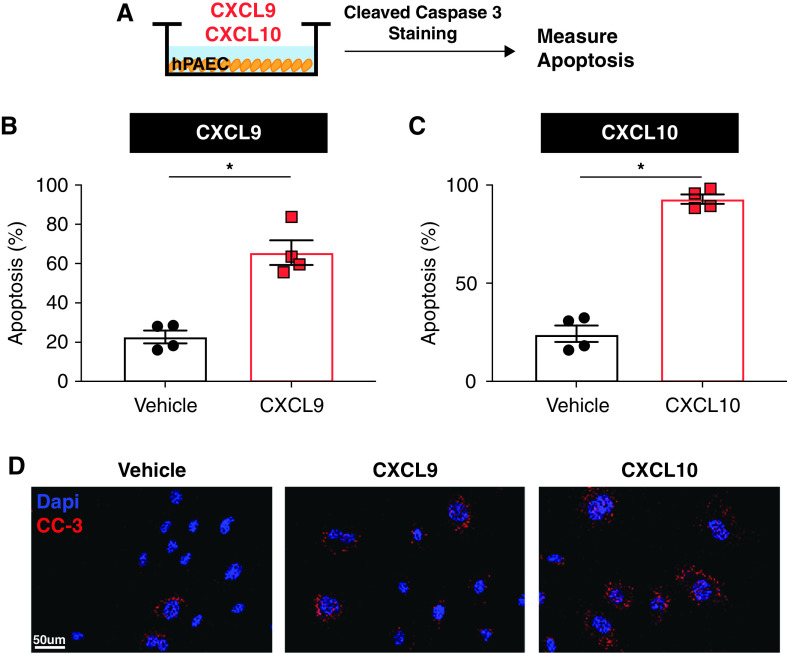
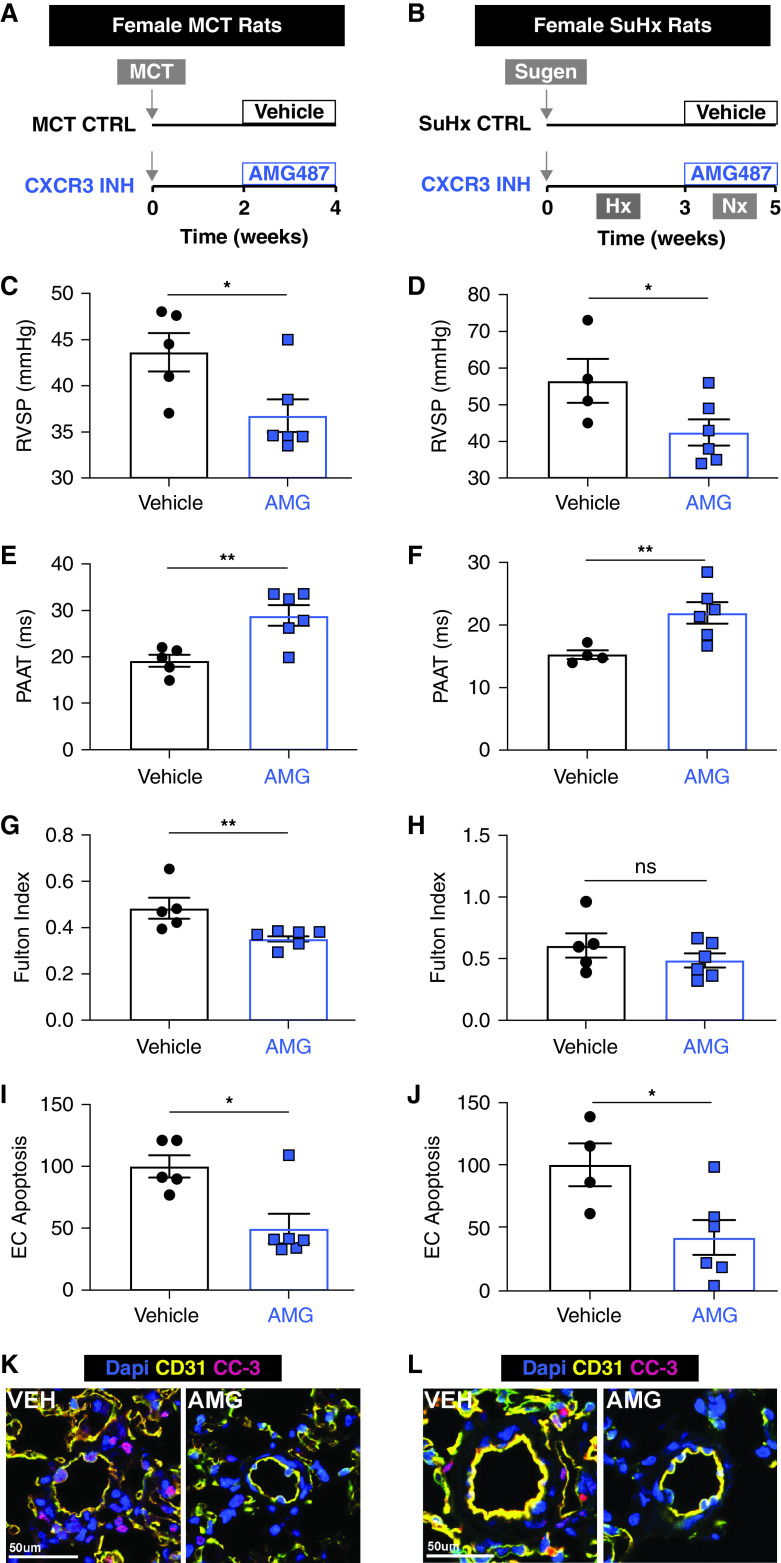
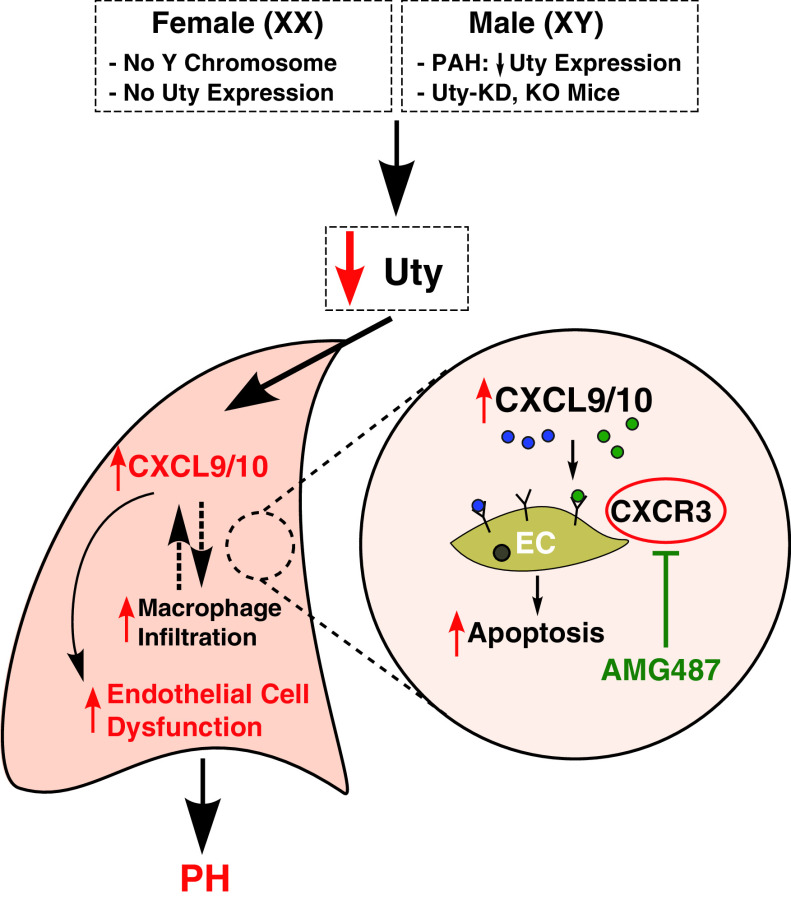
Rationale: Idiopathic pulmonary arterial hypertension (PAH) is a terminal pulmonary vascular disease characterized by increased pressure, right ventricular failure, and death. PAH exhibits a striking sex bias and is up to four times more prevalent in females. Understanding the molecular basis behind sex differences could help uncover novel therapies. Objectives: We previously discovered that the Y chromosome is protective against hypoxia-induced experimental pulmonary hypertension (PH), which may contribute to sex differences in PAH. Here, we identify the gene responsible for Y-chromosome protection, investigate key downstream autosomal genes, and demonstrate a novel preclinical therapy. Methods: To test the effect of Y-chromosome genes on PH development, we knocked down each Y-chromosome gene expressed in the lung by means of intratracheal instillation of siRNA in gonadectomized male mice exposed to hypoxia and monitored changes in right ventricular and pulmonary artery hemodynamics. We compared the lung transcriptome of Uty knockdown mouse lungs to those of male and female PAH patient lungs to identify common downstream pathogenic chemokines and tested the effects of these chemokines on human pulmonary artery endothelial cells. We further inhibited the activity of these chemokines in two preclinical pulmonary hypertension models to test the therapeutic efficacy. Measurements and Main Results: Knockdown of the Y-chromosome gene Uty resulted in more severe PH measured by increased right ventricular pressure and decreased pulmonary artery acceleration time. RNA sequencing revealed an increase in proinflammatory chemokines Cxcl9 and Cxcl10 as a result of Uty knockdown. We found CXCL9 and CXCL10 significantly upregulated in human PAH lungs, with more robust upregulation in females with PAH. Treatment of human pulmonary artery endothelial cells with CXCL9 and CXCL10 triggered apoptosis. Inhibition of Cxcl9 and Cxcl10 expression in male Uty knockout mice and CXCL9 and CXCL10 activity in female rats significantly reduced PH severity. Conclusions:Uty is protective against PH. Reduction of Uty expression results in increased expression of proinflammatory chemokines Cxcl9 and Cxcl10, which trigger endothelial cell death and PH. Inhibition of CLXC9 and CXLC10 rescues PH development in multiple experimental models.
Keywords: CXCL10; CXCL9; endothelial cells; pulmonary arterial hypertension; sex differences.
Figures
Comment in
-
A dUTY to Protect: Addressing "Y" We See Sex Differences in Pulmonary Hypertension.Am J Respir Crit Care Med. 2022 Jul 15;206(2):137-139. doi: 10.1164/rccm.202204-0653ED. Am J Respir Crit Care Med. 2022. PMID: 35549630 Free PMC article. No abstract available.
Similar articles
-
Single-Cell and Spatial Transcriptomics Identified Fatty Acid-Binding Proteins Controlling Endothelial Glycolytic and Arterial Programming in Pulmonary Hypertension.Arterioscler Thromb Vasc Biol. 2025 Jul;45(7):1145-1165. doi: 10.1161/ATVBAHA.124.321173. Epub 2025 May 22. Arterioscler Thromb Vasc Biol. 2025. PMID: 40401371 Free PMC article.
-
Endothelial Krüppel-like factor 4 modulates pulmonary arterial hypertension.Am J Respir Cell Mol Biol. 2014 Mar;50(3):647-53. doi: 10.1165/rcmb.2013-0135OC. Am J Respir Cell Mol Biol. 2014. PMID: 24156273 Free PMC article.
-
Selenoprotein P Promotes the Development of Pulmonary Arterial Hypertension: Possible Novel Therapeutic Target.Circulation. 2018 Aug 7;138(6):600-623. doi: 10.1161/CIRCULATIONAHA.117.033113. Circulation. 2018. PMID: 29636330
-
Lysine demethylases KDM6A and UTY: The X and Y of histone demethylation.Mol Genet Metab. 2019 May;127(1):31-44. doi: 10.1016/j.ymgme.2019.04.012. Epub 2019 May 6. Mol Genet Metab. 2019. PMID: 31097364 Review.
-
Therapeutic Approaches in Pulmonary Arterial Hypertension with Beneficial Effects on Right Ventricular Function-Preclinical Studies.Int J Mol Sci. 2023 Oct 24;24(21):15539. doi: 10.3390/ijms242115539. Int J Mol Sci. 2023. PMID: 37958522 Free PMC article. Review.
Cited by
-
Male-female comparisons are powerful in biomedical research - don't abandon them.Nature. 2024 May;629(8010):37-40. doi: 10.1038/d41586-024-01205-2. Nature. 2024. PMID: 38693409 No abstract available.
-
Role and Mechanism of Sialic Acid in Alleviating Acute Lung Injury through In Vivo and In Vitro Models.Foods. 2024 Sep 20;13(18):2984. doi: 10.3390/foods13182984. Foods. 2024. PMID: 39335912 Free PMC article.
-
Pulmonary Arterial Hypertension: Emerging Principles of Precision Medicine across Basic Science to Clinical Practice.Rev Cardiovasc Med. 2022;23(11):378. doi: 10.31083/j.rcm2311378. Epub 2022 Nov 9. Rev Cardiovasc Med. 2022. PMID: 36875282 Free PMC article.
-
Versatile JMJD proteins: juggling histones and much more.Trends Biochem Sci. 2024 Sep;49(9):804-818. doi: 10.1016/j.tibs.2024.06.009. Epub 2024 Jun 26. Trends Biochem Sci. 2024. PMID: 38926050 Free PMC article. Review.
-
Comprehensive transcriptomic analyses identify KDM genes-related subtypes with different TME infiltrates in gastric cancer.BMC Cancer. 2023 May 18;23(1):454. doi: 10.1186/s12885-023-10923-1. BMC Cancer. 2023. PMID: 37202737 Free PMC article.
References
-
- Walker AM, Langleben D, Korelitz JJ, Rich S, Rubin LJ, Strom BL, et al. Temporal trends and drug exposures in pulmonary hypertension: an American experience. Am Heart J . 2006;152:521–526. - PubMed
-
- Foderaro A, Ventetuolo CE. Pulmonary arterial hypertension and the sex hormone paradox. Curr Hypertens Rep . 2016;18:84. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
