

De novo-designed transmembrane domains tune engineered receptor functions
- PMID: 35506657
- PMCID: PMC9068223
- DOI: 10.7554/eLife.75660
De novo-designed transmembrane domains tune engineered receptor functions
Abstract
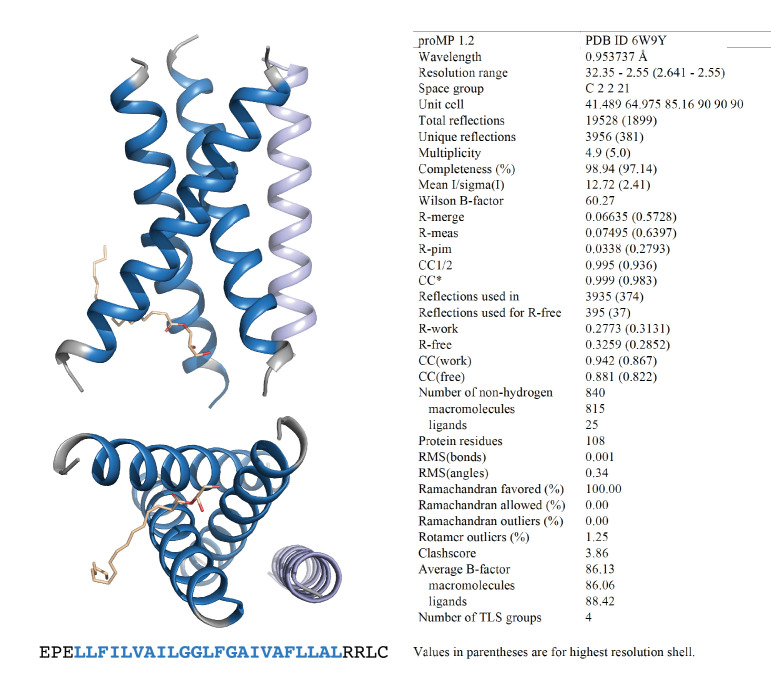
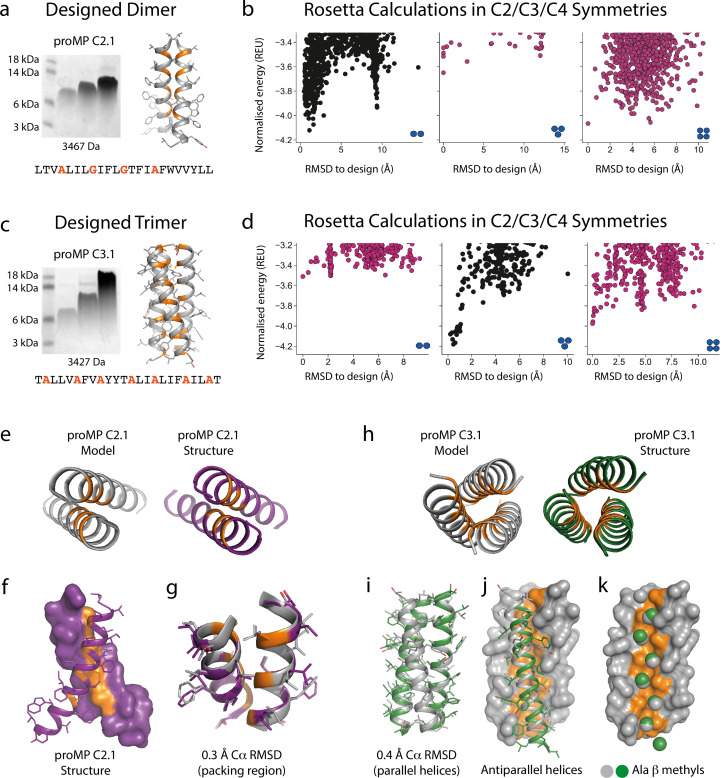
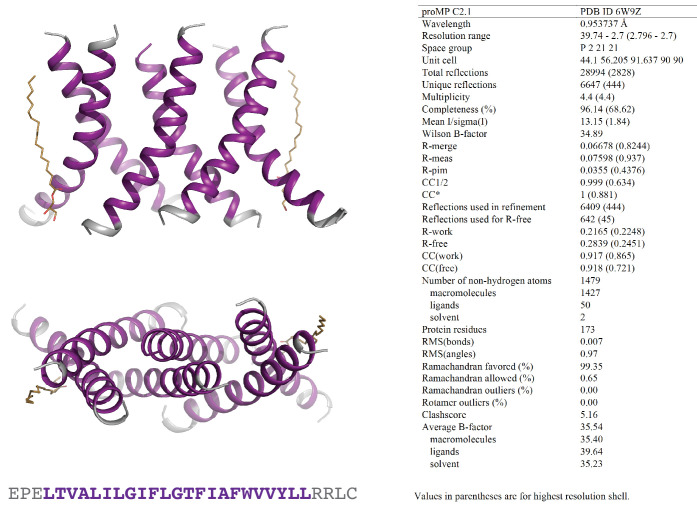
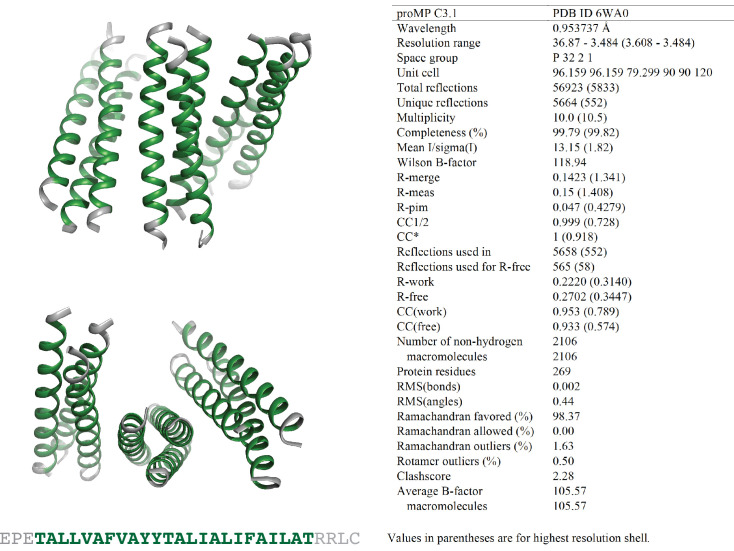
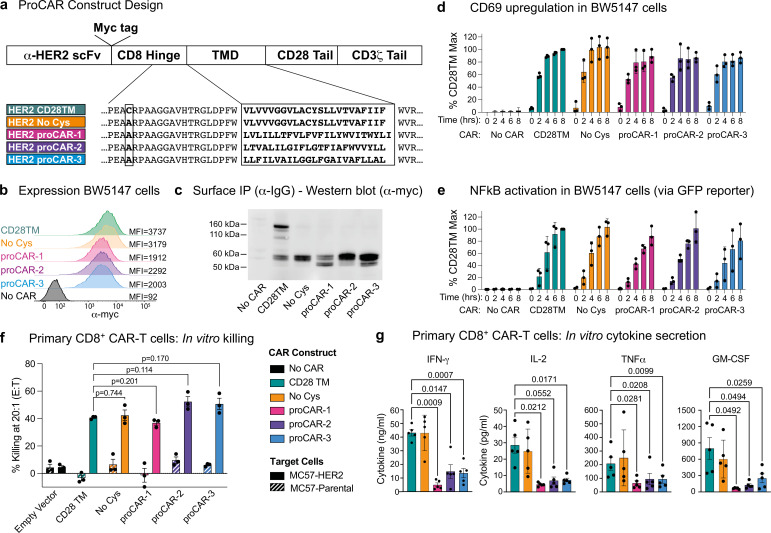
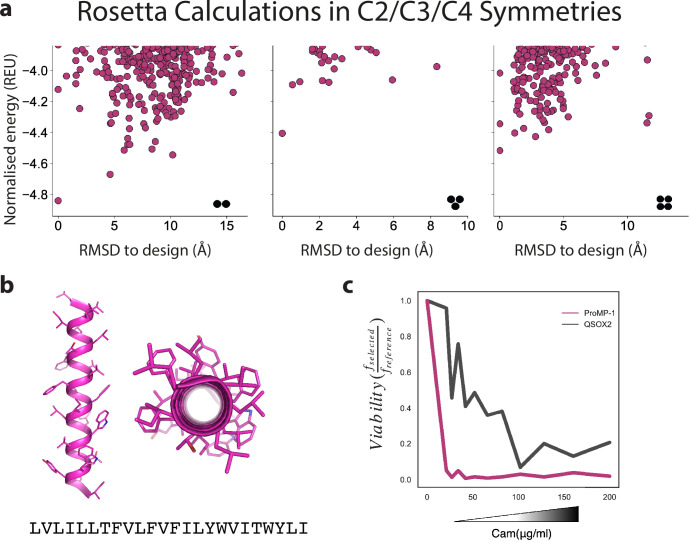
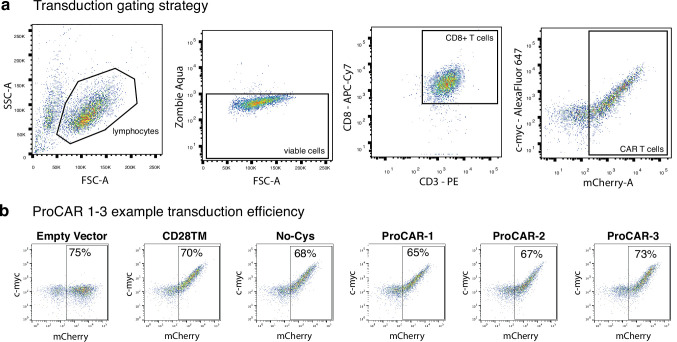
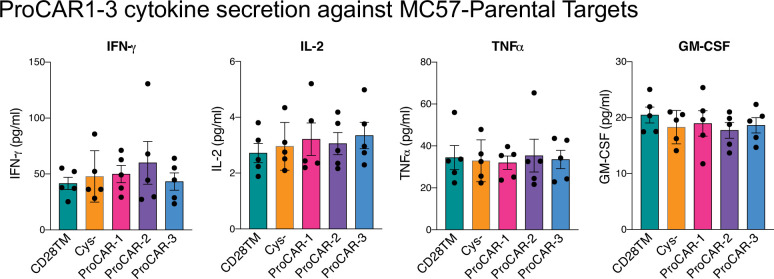
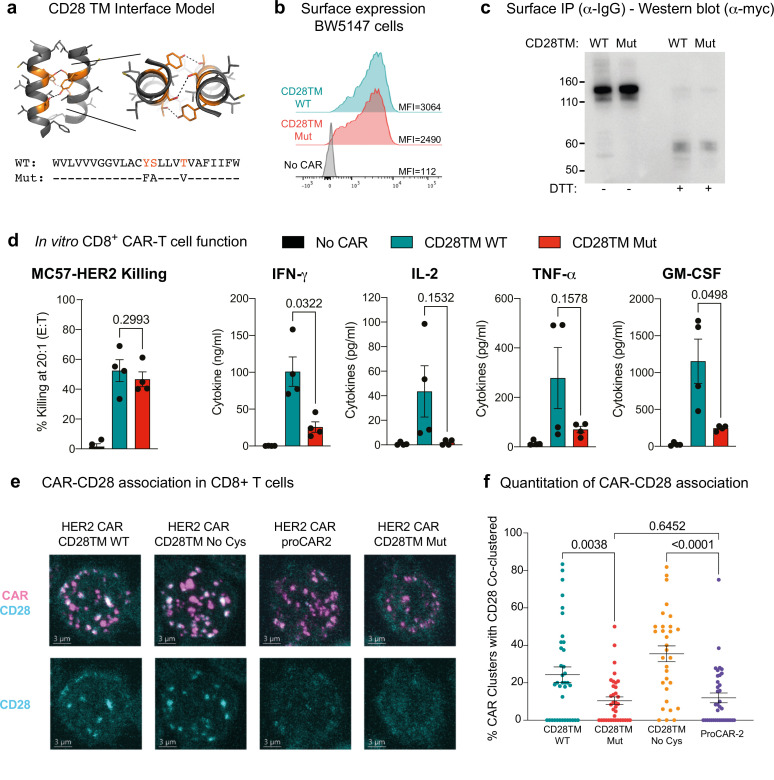
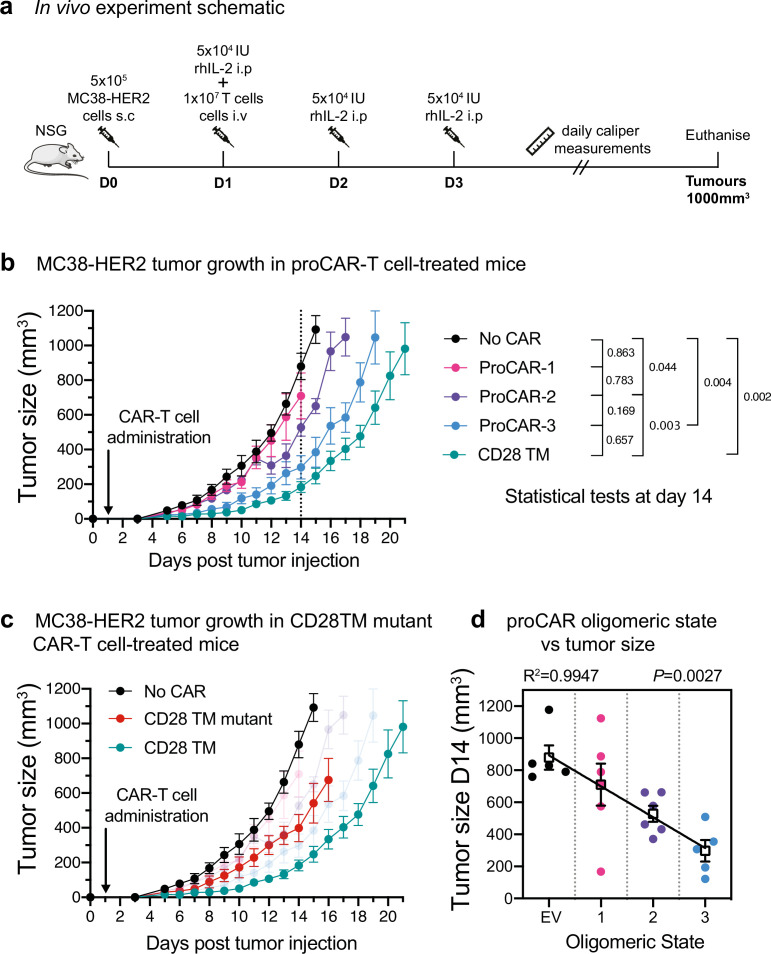
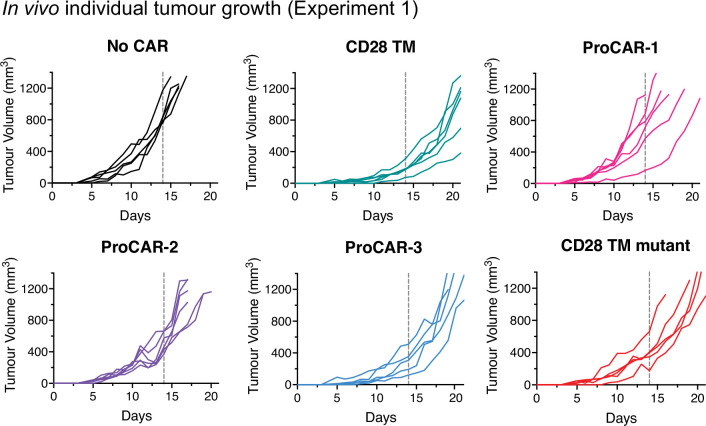
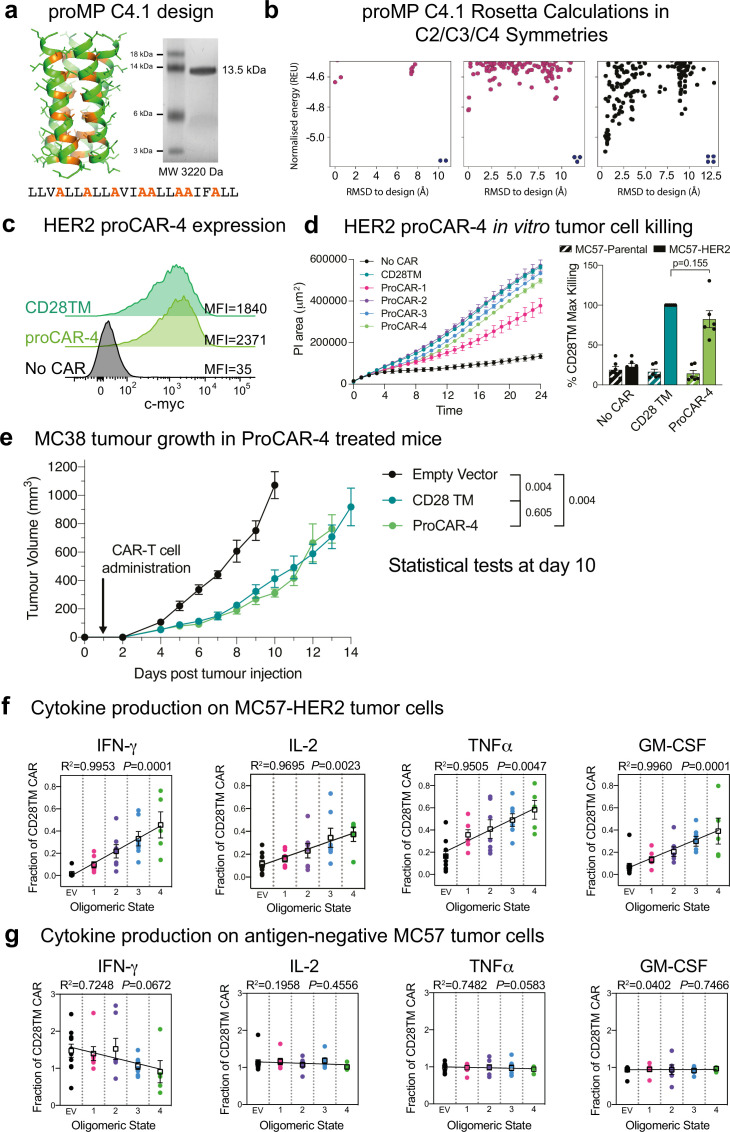
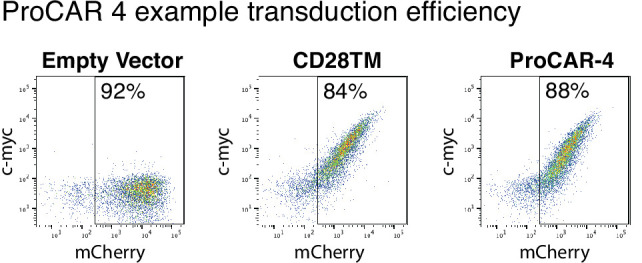
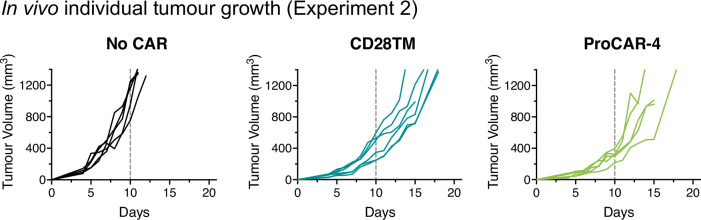
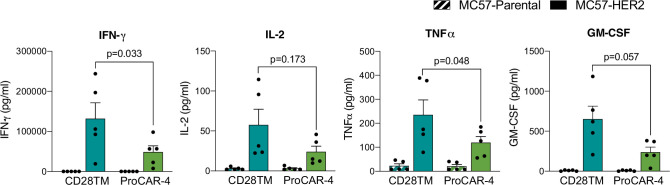
De novo-designed receptor transmembrane domains (TMDs) present opportunities for precise control of cellular receptor functions. We developed a de novo design strategy for generating programmed membrane proteins (proMPs): single-pass α-helical TMDs that self-assemble through computationally defined and crystallographically validated interfaces. We used these proMPs to program specific oligomeric interactions into a chimeric antigen receptor (CAR) that we expressed in mouse primary T cells and found that both in vitro CAR T cell cytokine release and in vivo antitumor activity scaled linearly with the oligomeric state encoded by the receptor TMD, from monomers up to tetramers. All programmed CARs stimulated substantially lower T cell cytokine release relative to the commonly used CD28 TMD, which we show elevated cytokine release through lateral recruitment of the endogenous T cell costimulatory receptor CD28. Precise design using orthogonal and modular TMDs thus provides a new way to program receptor structure and predictably tune activity for basic or applied synthetic biology.
Keywords: CAR T cell; E. coli; Rosetta; chimeric antigen receptor; de novo design; immunology; immunotherapy; inflammation; membrane protein; molecular biophysics; mouse; structural biology; transmembrane.
© 2022, Elazar et al.
Conflict of interest statement
AE, NC, JW, MC, MC Author is an inventor on a related patent WO2021229581A2, AD, JN, RT, RC, MJ No competing interests declared, SF Reviewing editor, eLife
Figures
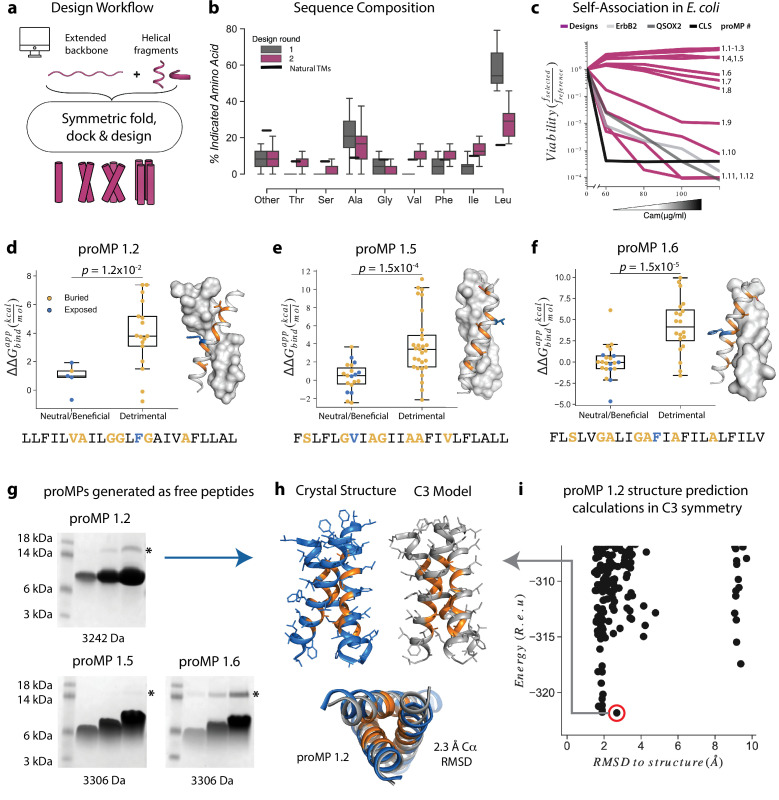
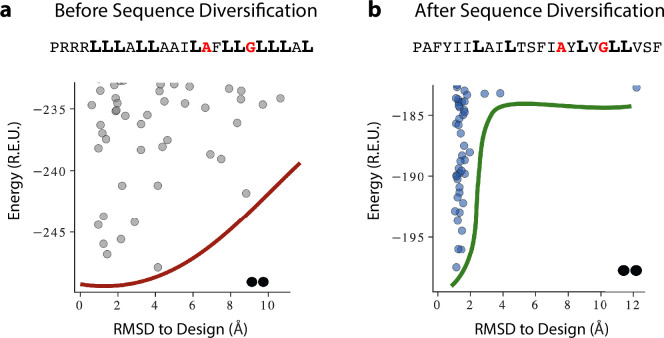
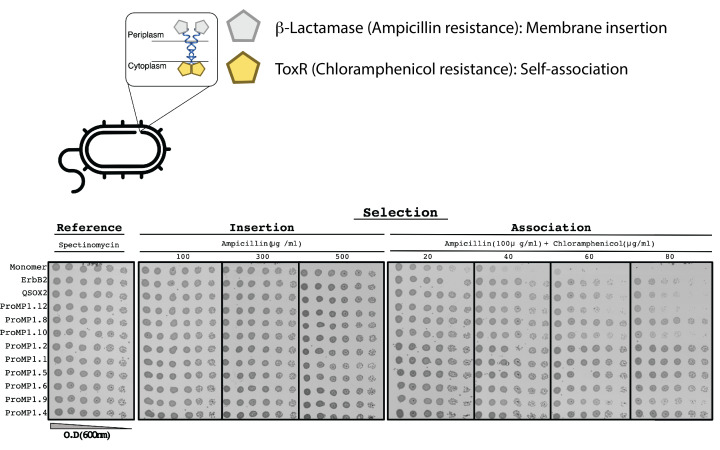
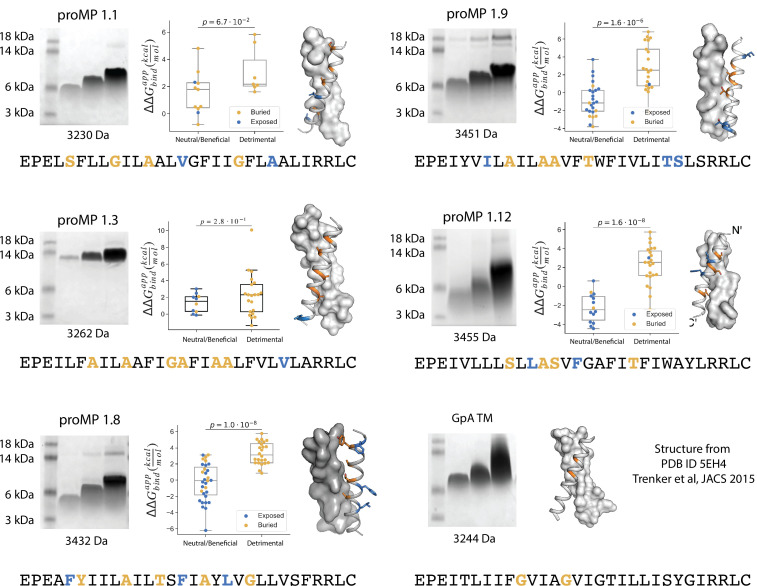
References
-
- Alabanza L, Pegues M, Geldres C, Shi V, Wiltzius JJW, Sievers SA, Yang S, Kochenderfer JN. Function of Novel Anti-CD19 Chimeric Antigen Receptors with Human Variable Regions Is Affected by Hinge and Transmembrane Domains. Molecular Therapy. 2017;25:2452–2465. doi: 10.1016/j.ymthe.2017.07.013. - DOI - PMC - PubMed
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Research Materials
