

Meta-imputation: An efficient method to combine genotype data after imputation with multiple reference panels
- PMID: 35508176
- PMCID: PMC9247833
- DOI: 10.1016/j.ajhg.2022.04.002
Meta-imputation: An efficient method to combine genotype data after imputation with multiple reference panels
Abstract
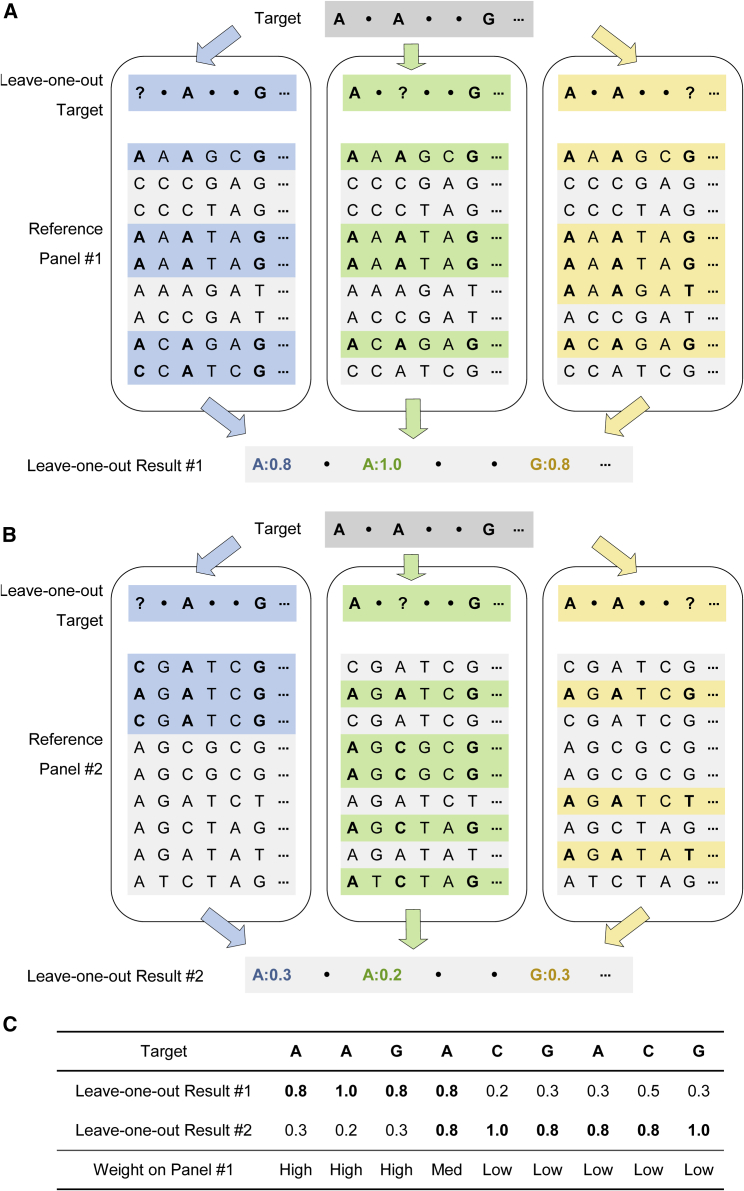
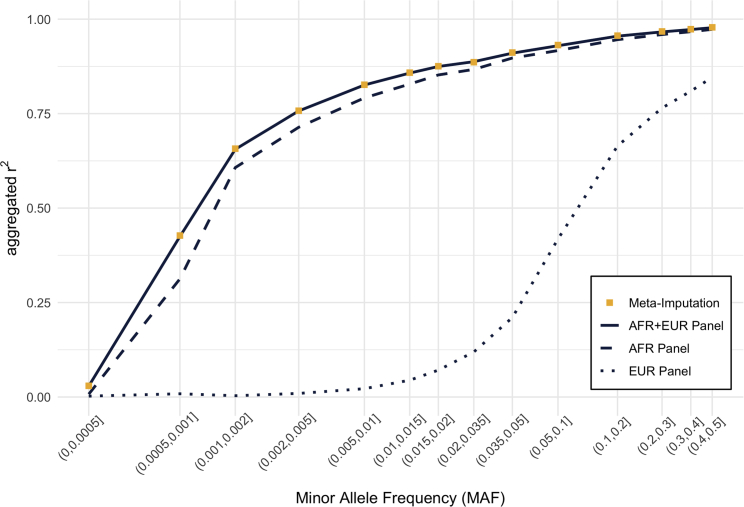
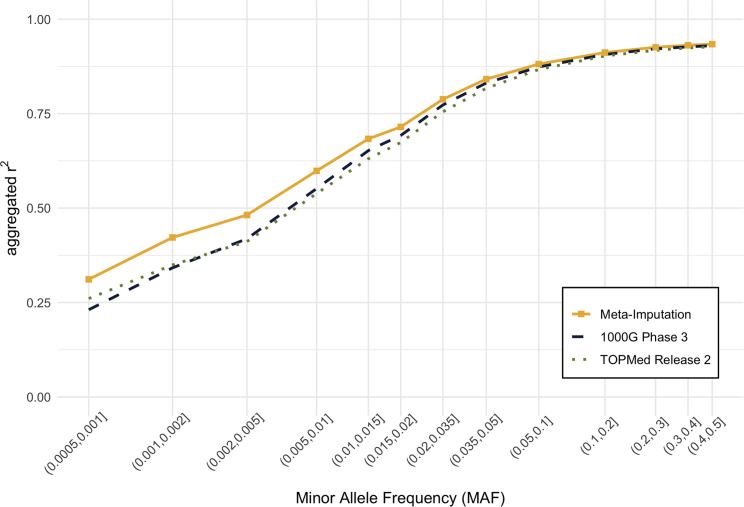
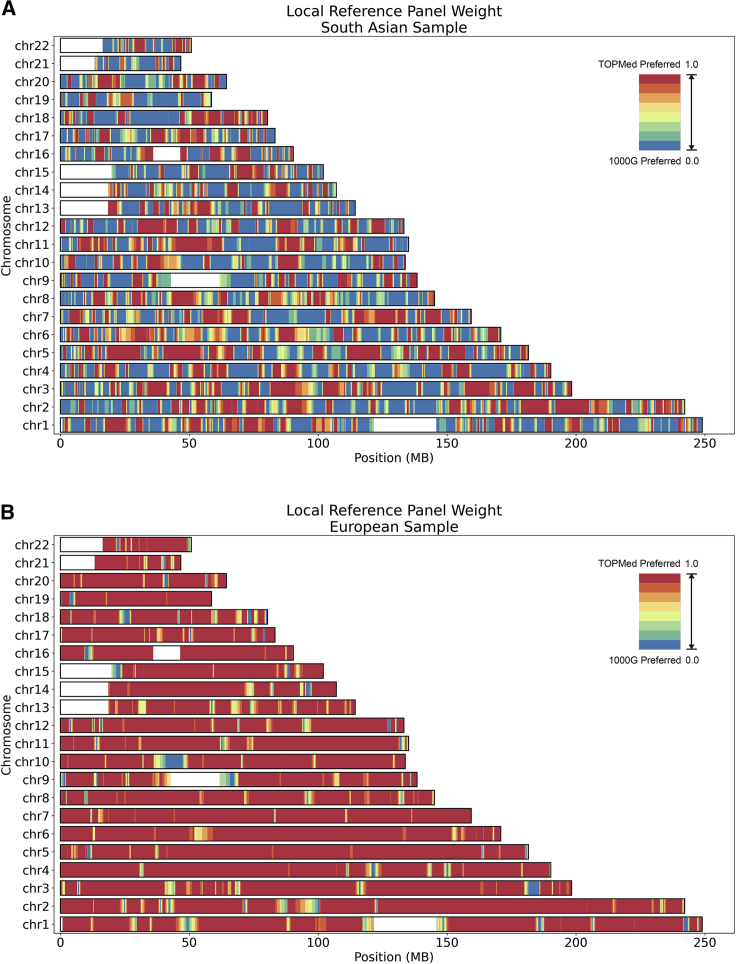
Genotype imputation is an integral tool in genome-wide association studies, in which it facilitates meta-analysis, increases power, and enables fine-mapping. With the increasing availability of whole-genome-sequence datasets, investigators have access to a multitude of reference-panel choices for genotype imputation. In principle, combining all sequenced whole genomes into a single large panel would provide the best imputation performance, but this is often cumbersome or impossible due to privacy restrictions. Here, we describe meta-imputation, a method that allows imputation results generated using different reference panels to be combined into a consensus imputed dataset. Our meta-imputation method requires small changes to the output of existing imputation tools to produce necessary inputs, which are then combined using dynamically estimated weights that are tailored to each individual and genome segment. In the scenarios we examined, the method consistently outperforms imputation using a single reference panel and achieves accuracy comparable to imputation using a combined reference panel.
Keywords: genome-wide association study; genotype imputation.
Copyright © 2022 American Society of Human Genetics. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests G.R.A. is an employee of Regeneron Pharmaceuticals and owns stock and stock options in Regeneron Pharmaceuticals.
Figures
References
-
- Fritsche L.G., Igl W., Bailey J.N.C., Grassmann F., Sengupta S., Bragg-Gresham J.L., Burdon K.P., Hebbring S.J., Wen C., Gorski M., et al. A large genome-wide association study of age-related macular degeneration highlights contributions of rare and common variants. Nat. Genet. 2016;48:134–143. doi: 10.1038/ng.3448. - DOI - PMC - PubMed
-
- Lee J.J., Wedow R., Okbay A., Kong E., Maghzian O., Zacher M., Nguyen-Viet T.A., Bowers P., Sidorenko J., Karlsson Linner R., et al. Gene discovery and polygenic prediction from a genome-wide association study of educational attainment in 1.1 million individuals. Nat. Genet. 2018;50:1112–1121. doi: 10.1038/s41588-018-0147-3. - DOI - PMC - PubMed
-
- Stahl E.A., Breen G., Forstner A.J., McQuillin A., Ripke S., Trubetskoy V., Mattheisen M., Wang Y., Coleman J.R.I., Gaspar H.A., et al. Genome-wide association study identifies 30 loci associated with bipolar disorder. Nat. Genet. 2019;51:793–803. doi: 10.1038/s41588-019-0397-8. - DOI - PMC - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
