

Therapeutic potential of deuterium-stabilized (R)-pioglitazone-PXL065-for X-linked adrenoleukodystrophy
- PMID: 35510808
- PMCID: PMC9545763
- DOI: 10.1002/jimd.12510
Therapeutic potential of deuterium-stabilized (R)-pioglitazone-PXL065-for X-linked adrenoleukodystrophy
Abstract
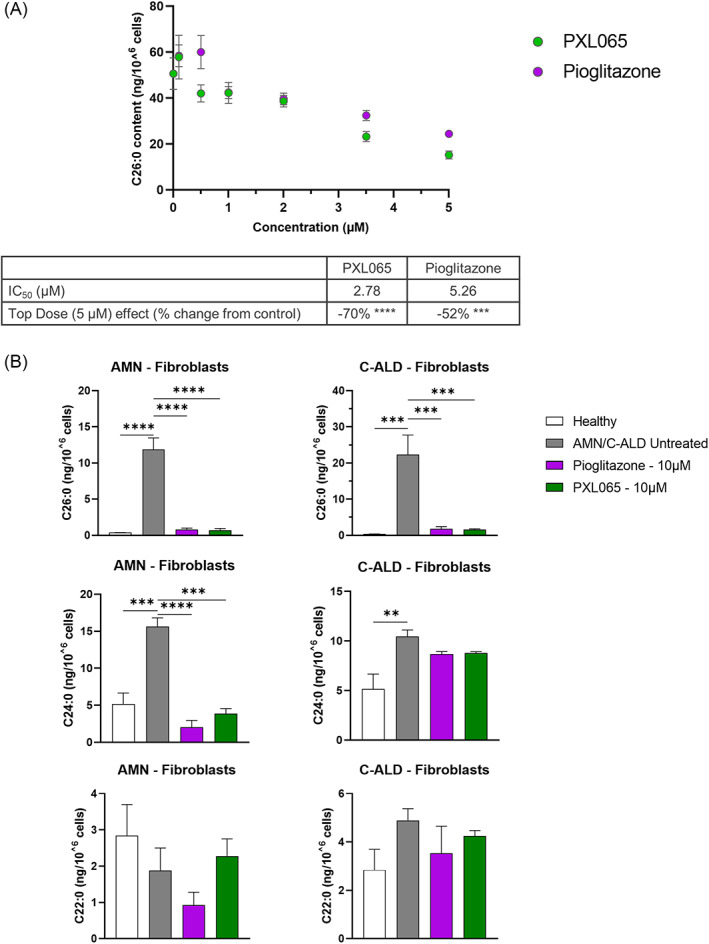
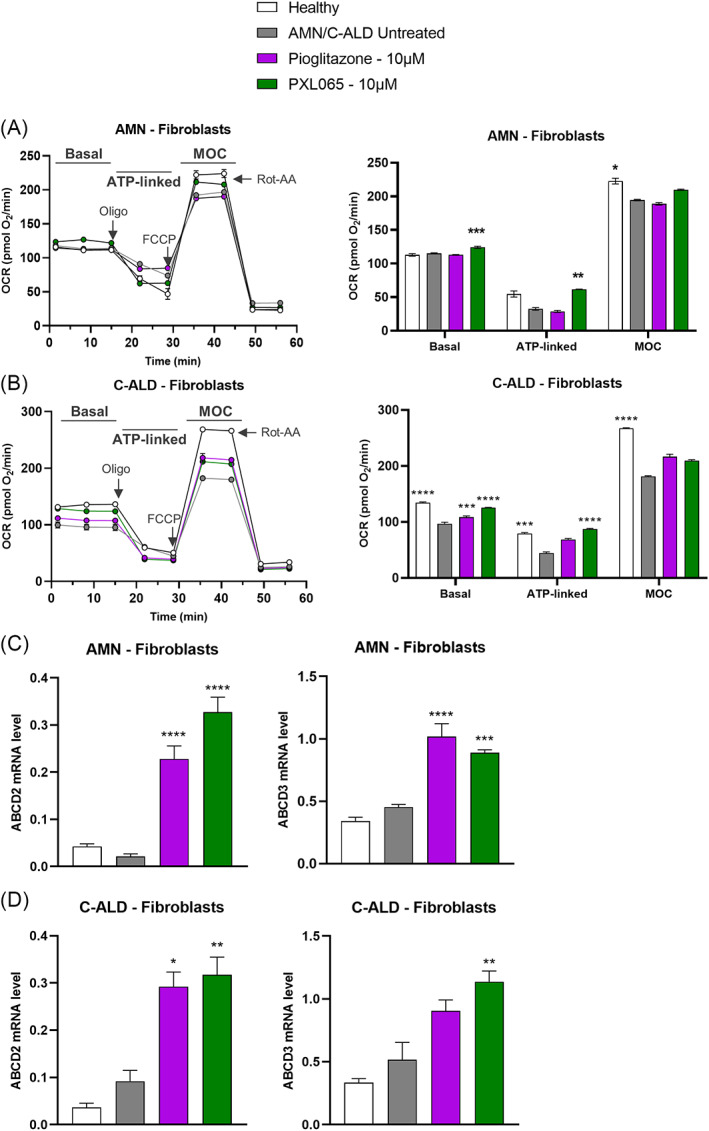
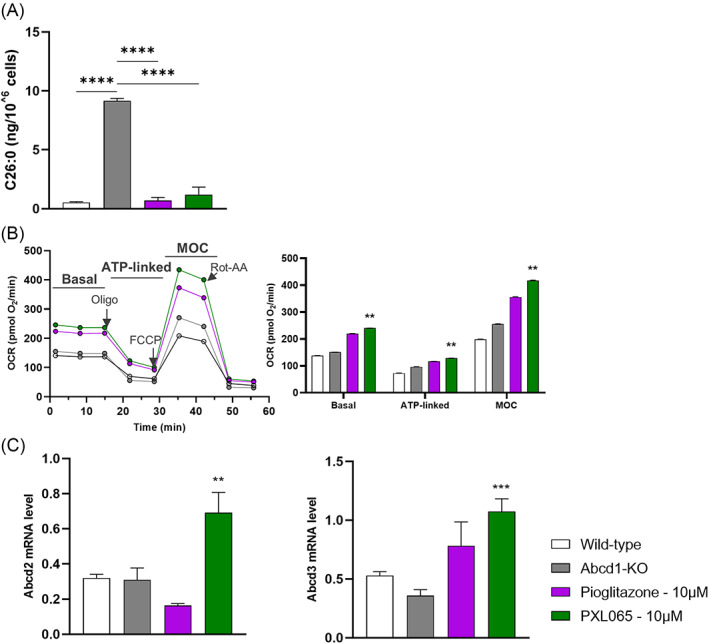
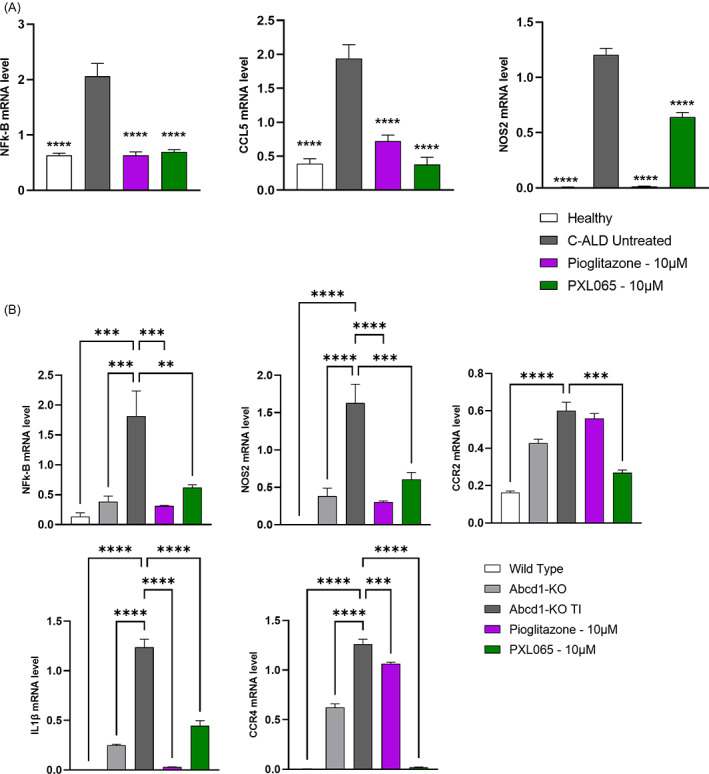
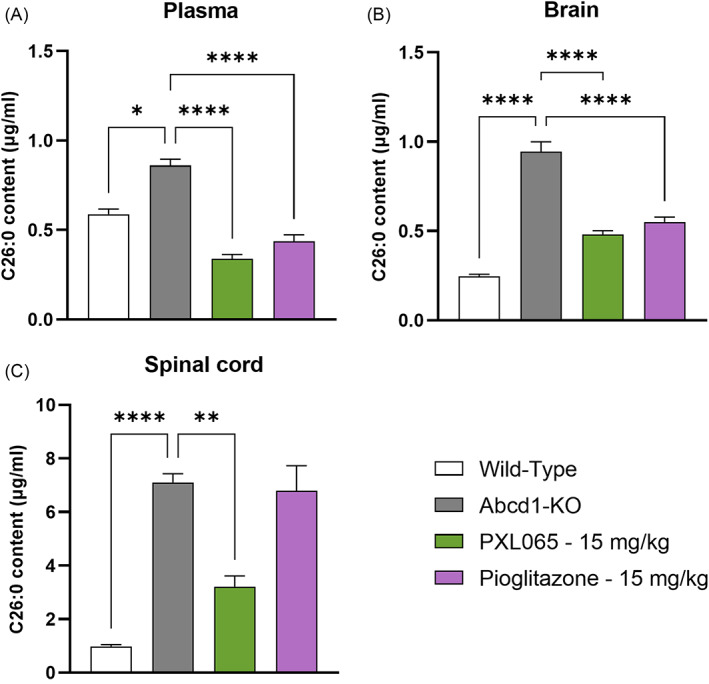
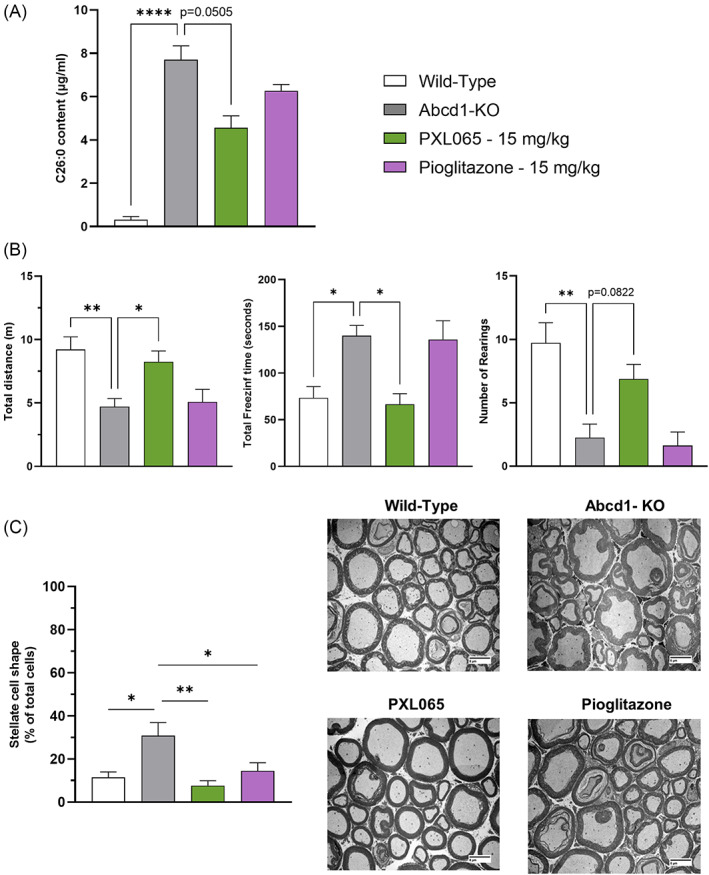
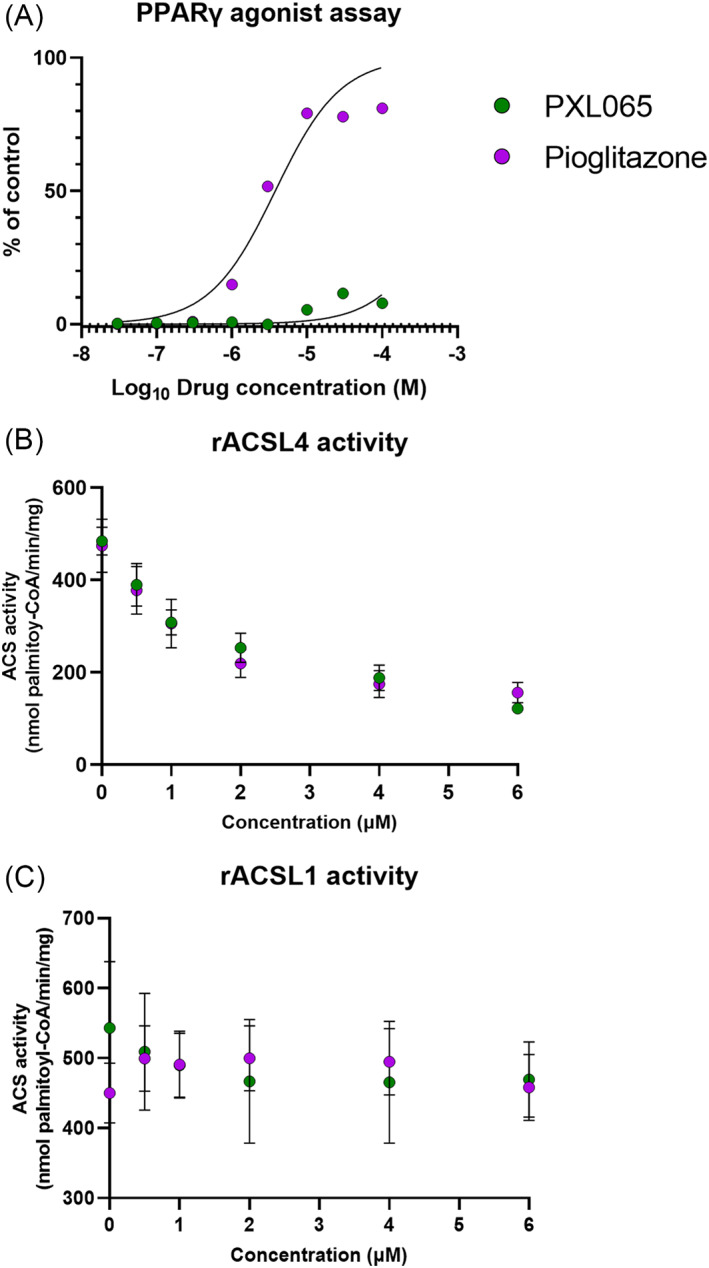
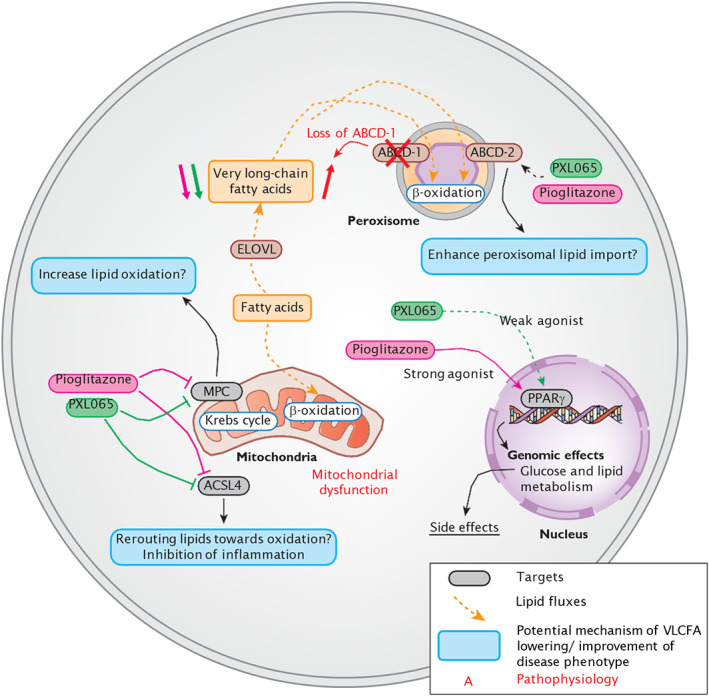
X-linked adrenoleukodystrophy (ALD) results from ABCD1 gene mutations which impair Very Long Chain Fatty Acids (VLCFA; C26:0 and C24:0) peroxisomal import and β-oxidation, leading to accumulation in plasma and tissues. Excess VLCFA drives impaired cellular functions (e.g. disrupted mitochondrial function), inflammation, and neurodegeneration. Major disease phenotypes include: adrenomyeloneuropathy (AMN), progressive spinal cord axonal degeneration, and cerebral ALD (C-ALD), inflammatory white matter demyelination and degeneration. No pharmacological treatment is available to-date for ALD. Pioglitazone, an anti-diabetic thiazolidinedione, exerts potential benefits in ALD models. Its mechanisms are genomic (PPARγ agonism) and nongenomic (mitochondrial pyruvate carrier-MPC, long-chain acyl-CoA synthetase 4-ACSL4, inhibition). However, its use is limited by PPARγ-driven side effects (e.g. weight gain, edema). PXL065 is a clinical-stage deuterium-stabilized (R)-enantiomer of pioglitazone which lacks PPARγ agonism but retains MPC activity. Here, we show that incubation of ALD patient-derived cells (both AMN and C-ALD) and glial cells from Abcd1-null mice with PXL065 resulted in: normalization of elevated VLCFA, improved mitochondrial function, and attenuated indices of inflammation. Compensatory peroxisomal transporter gene expression was also induced. Additionally, chronic treatment of Abcd1-null mice lowered VLCFA in plasma, brain and spinal cord and improved both neural histology (sciatic nerve) and neurobehavioral test performance. Several in vivo effects of PXL065 exceeded those achieved with pioglitazone. PXL065 was confirmed to lack PPARγ agonism but retained ACSL4 activity of pioglitazone. PXL065 has novel actions and mechanisms and exhibits a range of potential benefits in ALD models; further testing of this molecule in ALD patients is warranted.
© 2022 Poxel SA. Journal of Inherited Metabolic Disease published by John Wiley & Sons Ltd on behalf of SSIEM.
Conflict of interest statement
Pierre‐Axel Monternier is an employee and shareholder of Poxel. Jaspreet Singh received sponsored research grants from Poxel to support the conduct of experiments related to the aims of this manuscript and is also a member of Poxel's Scientific Advisory Board. Parveen Parasar declares no conflict of interest. Pierre Theurey is an employee and shareholder of Poxel. Sheila DeWitt received sponsored research grants from Poxel to support the conduct of experiments related to the aims of this manuscript. Vincent Jacques received sponsored research grants from Poxel to support the conduct of experiments related to the aims of this manuscript. Eric Klett received sponsored research grants from Poxel to support the conduct of experiments related to the aims of this manuscript. Navtej Kaur declares no conflict of interest. Tavarekere N. Nagaraja declares no conflict of interest. David E. Moller is an employee and shareholder of Poxel. Sophie Hallakou‐Bozec is an employee and shareholder of Poxel.
Figures
References
-
- Moser HW, Mahmood A, Raymond GV. X‐linked adrenoleukodystrophy. Nat Clin Pract Neurol. 2007;3(3):140‐151. - PubMed
-
- Moser HW, Moser AB, Frayer KK, et al. Adrenoleukodystrophy. Increased plasma content of saturated very long chain fatty acids. Neurology. 1981;31(10):1241. - PubMed
-
- Eichler FS, Ren JQ, Cossoy M, et al. Is microglial apoptosis an early pathogenic change in cerebral X‐linked adrenoleukodystrophy? Ann Neurol. 2008;63(6):729‐742. - PubMed
-
- Hein S, Schonfeld P, Kahlert S, Reiser G. Toxic effects of X‐linked adrenoleukodystrophy‐associated, very long chain fatty acids on glial cells and neurons from rat hippocampus in culture. Hum Mol Genet. 2008;17(12):1750‐1761. - PubMed
-
- Fourcade S, Lopez‐Erauskin J, Galino J, et al. Early oxidative damage underlying neurodegeneration in X‐adrenoleukodystrophy. Hum Mol Genet. 2008;17(12):1762‐1773. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Research Materials
