

Genetic Analysis of Lung Cancer and the Germline Impact on Somatic Mutation Burden
- PMID: 35511172
- PMCID: PMC9360465
- DOI: 10.1093/jnci/djac087
Genetic Analysis of Lung Cancer and the Germline Impact on Somatic Mutation Burden
Abstract
Background: Germline genetic variation contributes to lung cancer (LC) susceptibility. Previous genome-wide association studies (GWAS) have implicated susceptibility loci involved in smoking behaviors and DNA repair genes, but further work is required to identify susceptibility variants.
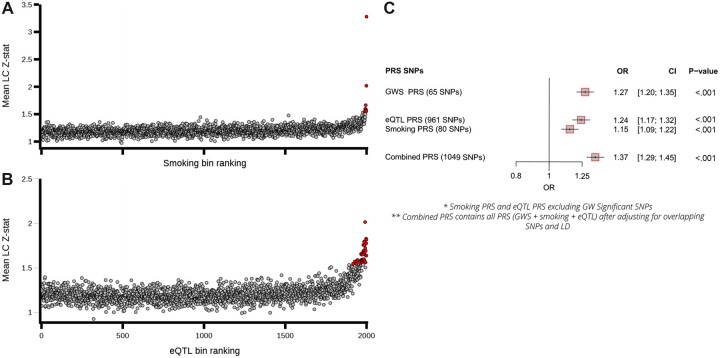
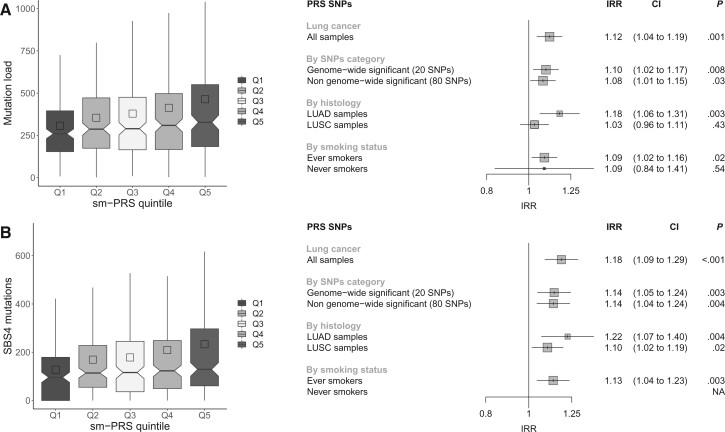
Methods: To identify LC susceptibility loci, a family history-based genome-wide association by proxy (GWAx) of LC (48 843 European proxy LC patients, 195 387 controls) was combined with a previous LC GWAS (29 266 patients, 56 450 controls) by meta-analysis. Colocalization was used to explore candidate genes and overlap with existing traits at discovered susceptibility loci. Polygenic risk scores (PRS) were tested within an independent validation cohort (1 666 LC patients vs 6 664 controls) using variants selected from the LC susceptibility loci and a novel selection approach using published GWAS summary statistics. Finally, the effects of the LC PRS on somatic mutational burden were explored in patients whose tumor resections have been profiled by exome (n = 685) and genome sequencing (n = 61). Statistical tests were 2-sided.
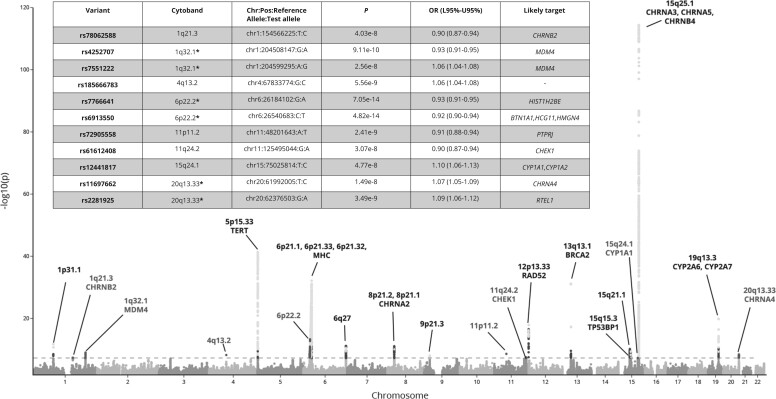
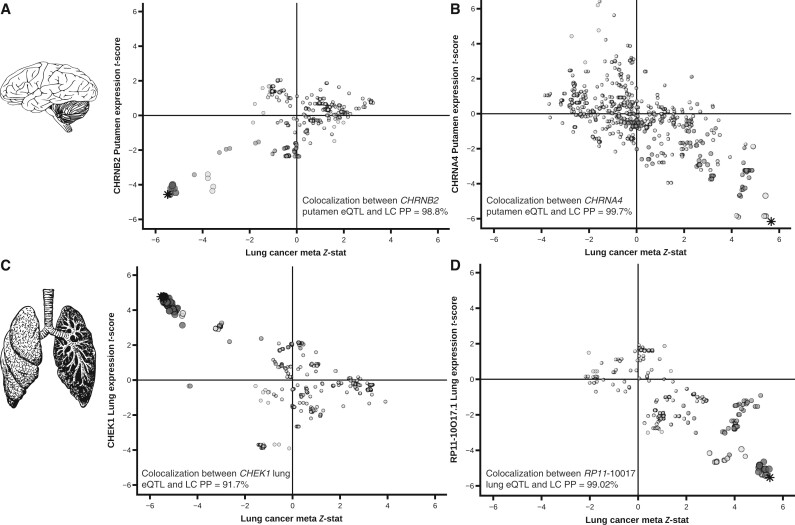
Results: The GWAx-GWAS meta-analysis identified 8 novel LC loci. Colocalization implicated DNA repair genes (CHEK1), metabolic genes (CYP1A1), and smoking propensity genes (CHRNA4 and CHRNB2). PRS analysis demonstrated that these variants, as well as subgenome-wide significant variants related to expression quantitative trait loci and/or smoking propensity, assisted in LC genetic risk prediction (odds ratio = 1.37, 95% confidence interval = 1.29 to 1.45; P < .001). Patients with higher genetic PRS loads of smoking-related variants tended to have higher mutation burdens in their lung tumors.
Conclusions: This study has expanded the number of LC susceptibility loci and provided insights into the molecular mechanisms by which these susceptibility variants contribute to LC development.
© The Author(s) 2022. Published by Oxford University Press.
Figures
References
-
- Tokuhata GK, Lilienfeld AM.. Familial aggregation of lung cancer in humans. J Natl Cancer Inst. 1963;30(2):289-312. - PubMed
