

Anti-ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP-mutant ACVR1
- PMID: 35511419
- PMCID: PMC9197526
- DOI: 10.1172/JCI153792
Anti-ACVR1 antibodies exacerbate heterotopic ossification in fibrodysplasia ossificans progressiva (FOP) by activating FOP-mutant ACVR1
Abstract
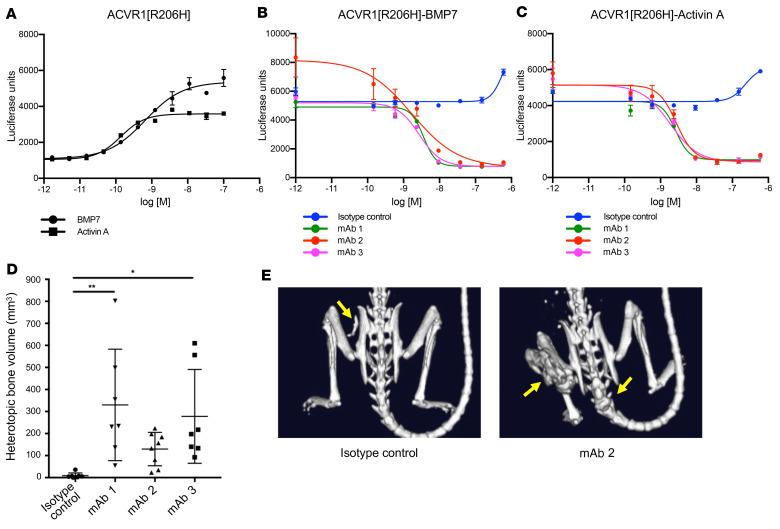
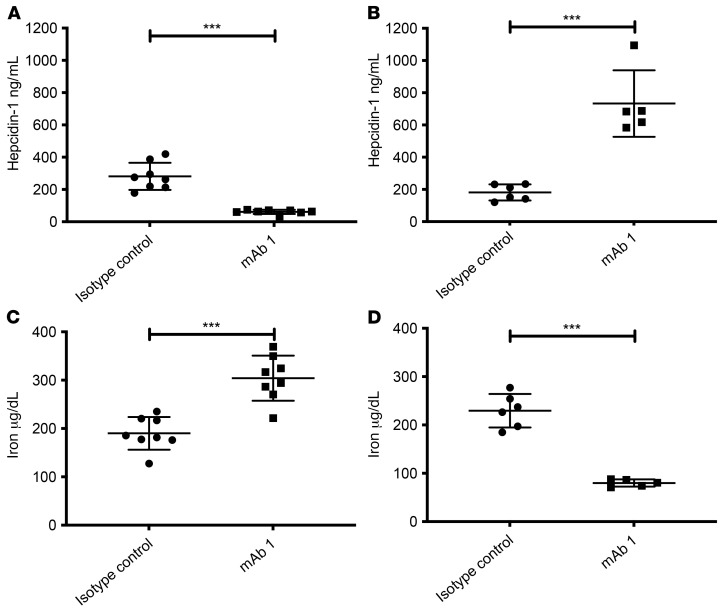
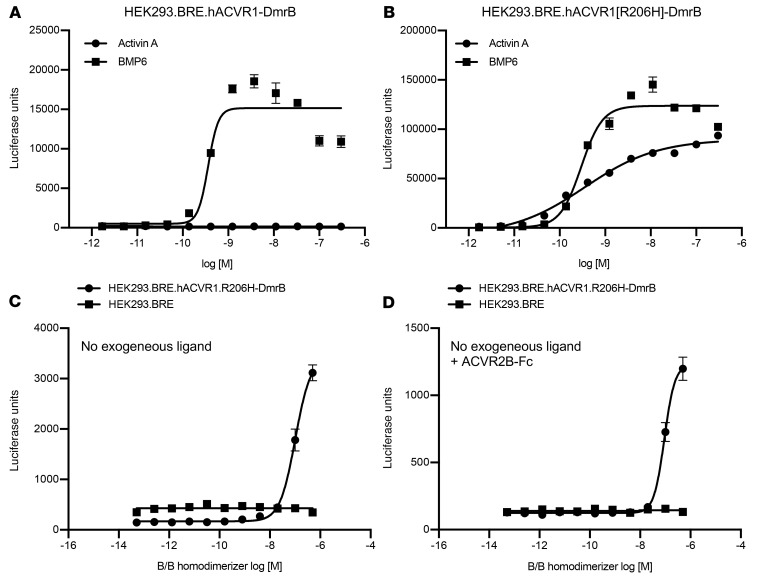
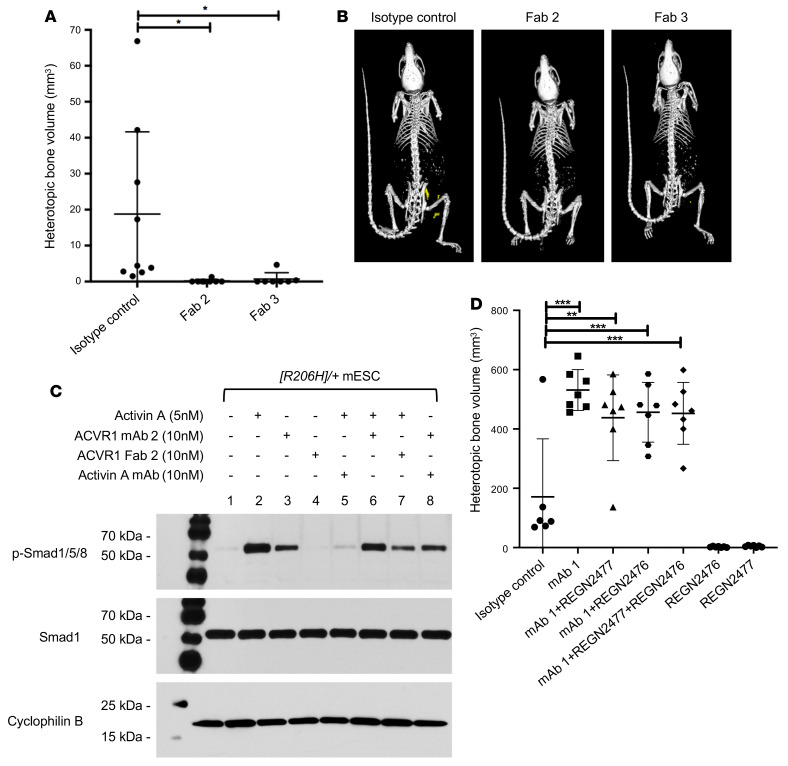
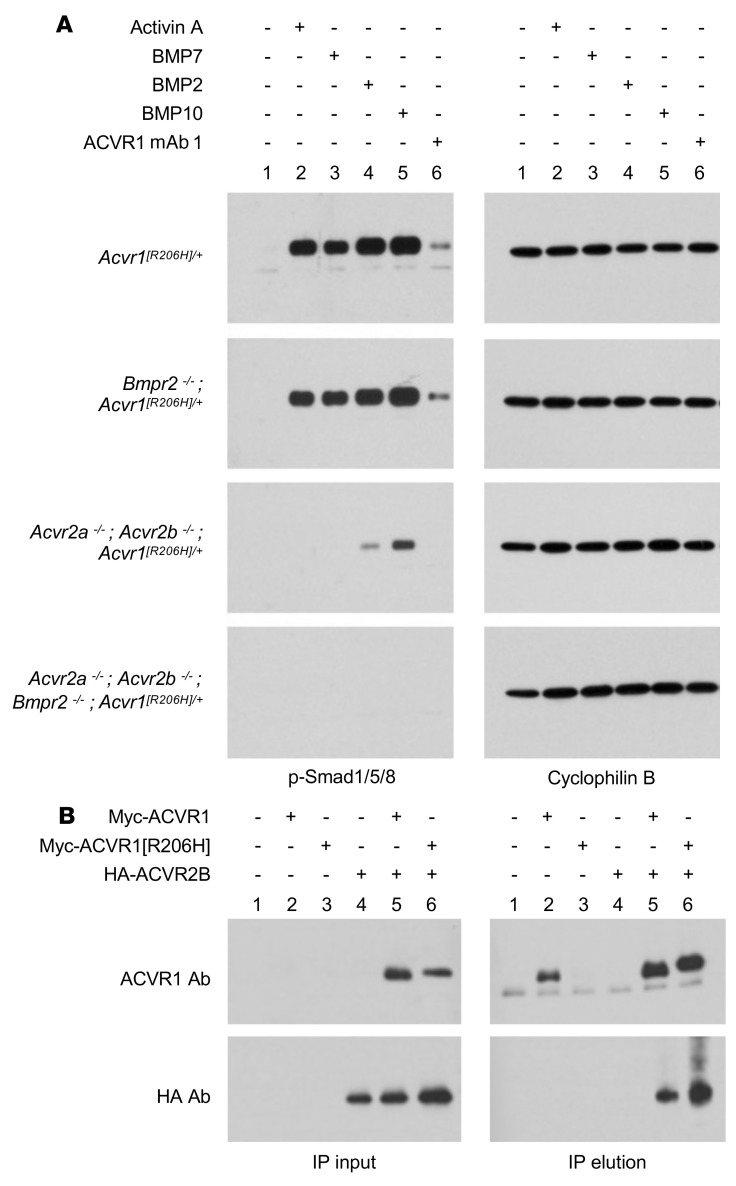
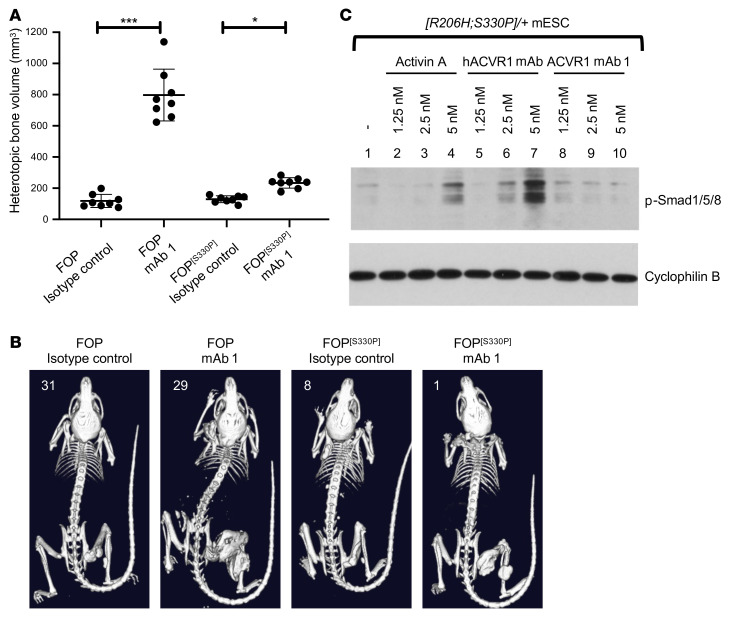
Fibrodysplasia ossificans progressiva (FOP) is a rare genetic disorder whose most debilitating pathology is progressive and cumulative heterotopic ossification (HO) of skeletal muscles, ligaments, tendons, and fascia. FOP is caused by mutations in the type I BMP receptor gene ACVR1, which enable ACVR1 to utilize its natural antagonist, activin A, as an agonistic ligand. The physiological relevance of this property is underscored by the fact that HO in FOP is exquisitely dependent on activation of FOP-mutant ACVR1 by activin A, an effect countered by inhibition of anti-activin A via monoclonal antibody treatment. Hence, we surmised that anti-ACVR1 antibodies that block activation of ACVR1 by ligands should also inhibit HO in FOP and provide an additional therapeutic option for this condition. Therefore, we generated anti-ACVR1 monoclonal antibodies that block ACVR1's activation by its ligands. Surprisingly, in vivo, these anti-ACVR1 antibodies stimulated HO and activated signaling of FOP-mutant ACVR1. This property was restricted to FOP-mutant ACVR1 and resulted from anti-ACVR1 antibody-mediated dimerization of ACVR1. Conversely, wild-type ACVR1 was inhibited by anti-ACVR1 antibodies. These results uncover an additional property of FOP-mutant ACVR1 and indicate that anti-ACVR1 antibodies should not be considered as therapeutics for FOP.
Keywords: Bone Biology; Bone disease; Genetic diseases; Signal transduction; Therapeutics.
Figures
Comment in
-
Twists in the fibrodysplasia ossificans progressiva story challenge and expand our understanding of BMP biology.J Clin Invest. 2022 Jun 15;132(12):e160773. doi: 10.1172/JCI160773. J Clin Invest. 2022. PMID: 35703179 Free PMC article.
Similar articles
-
Overexpression of Wild-Type ACVR1 in Fibrodysplasia Ossificans Progressiva Mice Rescues Perinatal Lethality and Inhibits Heterotopic Ossification.J Bone Miner Res. 2022 Nov;37(11):2077-2093. doi: 10.1002/jbmr.4617. Epub 2022 Jul 3. J Bone Miner Res. 2022. PMID: 35637634 Free PMC article.
-
The obligatory role of Activin A in the formation of heterotopic bone in Fibrodysplasia Ossificans Progressiva.Bone. 2018 Apr;109:210-217. doi: 10.1016/j.bone.2017.06.011. Epub 2017 Jun 16. Bone. 2018. PMID: 28629737 Free PMC article. Review.
-
The ACVR1 R206H mutation found in fibrodysplasia ossificans progressiva increases human induced pluripotent stem cell-derived endothelial cell formation and collagen production through BMP-mediated SMAD1/5/8 signaling.Stem Cell Res Ther. 2016 Aug 17;7(1):115. doi: 10.1186/s13287-016-0372-6. Stem Cell Res Ther. 2016. PMID: 27530160 Free PMC article.
-
Depletion of Mast Cells and Macrophages Impairs Heterotopic Ossification in an Acvr1R206H Mouse Model of Fibrodysplasia Ossificans Progressiva.J Bone Miner Res. 2018 Feb;33(2):269-282. doi: 10.1002/jbmr.3304. Epub 2018 Jan 3. J Bone Miner Res. 2018. PMID: 28986986 Free PMC article.
-
How Activin A Became a Therapeutic Target in Fibrodysplasia Ossificans Progressiva.Biomolecules. 2024 Jan 12;14(1):101. doi: 10.3390/biom14010101. Biomolecules. 2024. PMID: 38254701 Free PMC article. Review.
Cited by
-
Sex as a Critical Variable in Basic and Pre-Clinical Studies of Fibrodysplasia Ossificans Progressiva.Biomolecules. 2024 Feb 1;14(2):177. doi: 10.3390/biom14020177. Biomolecules. 2024. PMID: 38397414 Free PMC article.
-
Overexpression of Wild-Type ACVR1 in Fibrodysplasia Ossificans Progressiva Mice Rescues Perinatal Lethality and Inhibits Heterotopic Ossification.J Bone Miner Res. 2022 Nov;37(11):2077-2093. doi: 10.1002/jbmr.4617. Epub 2022 Jul 3. J Bone Miner Res. 2022. PMID: 35637634 Free PMC article.
-
Fibrodysplasia ossificans progressiva: genetic and clinical characterization in a cohort of Polish patients and review of potential therapies.J Appl Genet. 2025 Apr 12. doi: 10.1007/s13353-025-00966-4. Online ahead of print. J Appl Genet. 2025. PMID: 40220125
-
Heterotopic ossification: Current developments and emerging potential therapies.Chin Med J (Engl). 2025 Feb 20;138(4):389-404. doi: 10.1097/CM9.0000000000003244. Epub 2025 Jan 17. Chin Med J (Engl). 2025. PMID: 39819765 Free PMC article. Review.
-
Reduced GS Domain Serine/Threonine Requirements of Fibrodysplasia Ossificans Progressiva Mutant Type I BMP Receptor ACVR1 in the Zebrafish.J Bone Miner Res. 2023 Sep;38(9):1364-1385. doi: 10.1002/jbmr.4869. Epub 2023 Jul 17. J Bone Miner Res. 2023. PMID: 37329499 Free PMC article.
References
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Other Literature Sources
