

AliSim: A Fast and Versatile Phylogenetic Sequence Simulator for the Genomic Era
- PMID: 35511713
- PMCID: PMC9113491
- DOI: 10.1093/molbev/msac092
AliSim: A Fast and Versatile Phylogenetic Sequence Simulator for the Genomic Era
Abstract
Sequence simulators play an important role in phylogenetics. Simulated data has many applications, such as evaluating the performance of different methods, hypothesis testing with parametric bootstraps, and, more recently, generating data for training machine-learning applications. Many sequence simulation programmes exist, but the most feature-rich programmes tend to be rather slow, and the fastest programmes tend to be feature-poor. Here, we introduce AliSim, a new tool that can efficiently simulate biologically realistic alignments under a large range of complex evolutionary models. To achieve high performance across a wide range of simulation conditions, AliSim implements an adaptive approach that combines the commonly used rate matrix and probability matrix approaches. AliSim takes 1.4 h and 1.3 GB RAM to simulate alignments with one million sequences or sites, whereas popular software Seq-Gen, Dawg, and INDELible require 2-5 h and 50-500 GB of RAM. We provide AliSim as an extension of the IQ-TREE software version 2.2, freely available at www.iqtree.org, and a comprehensive user tutorial at http://www.iqtree.org/doc/AliSim.
Keywords: molecular evolution; phylogenetics; sequence simulation.
© The Author(s) 2022. Published by Oxford University Press on behalf of Society for Molecular Biology and Evolution.
Figures
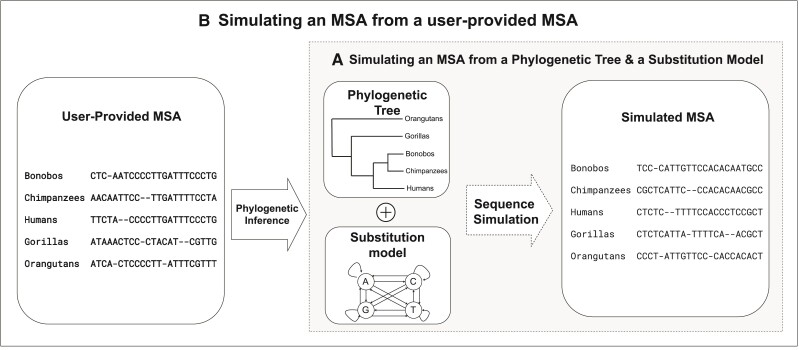
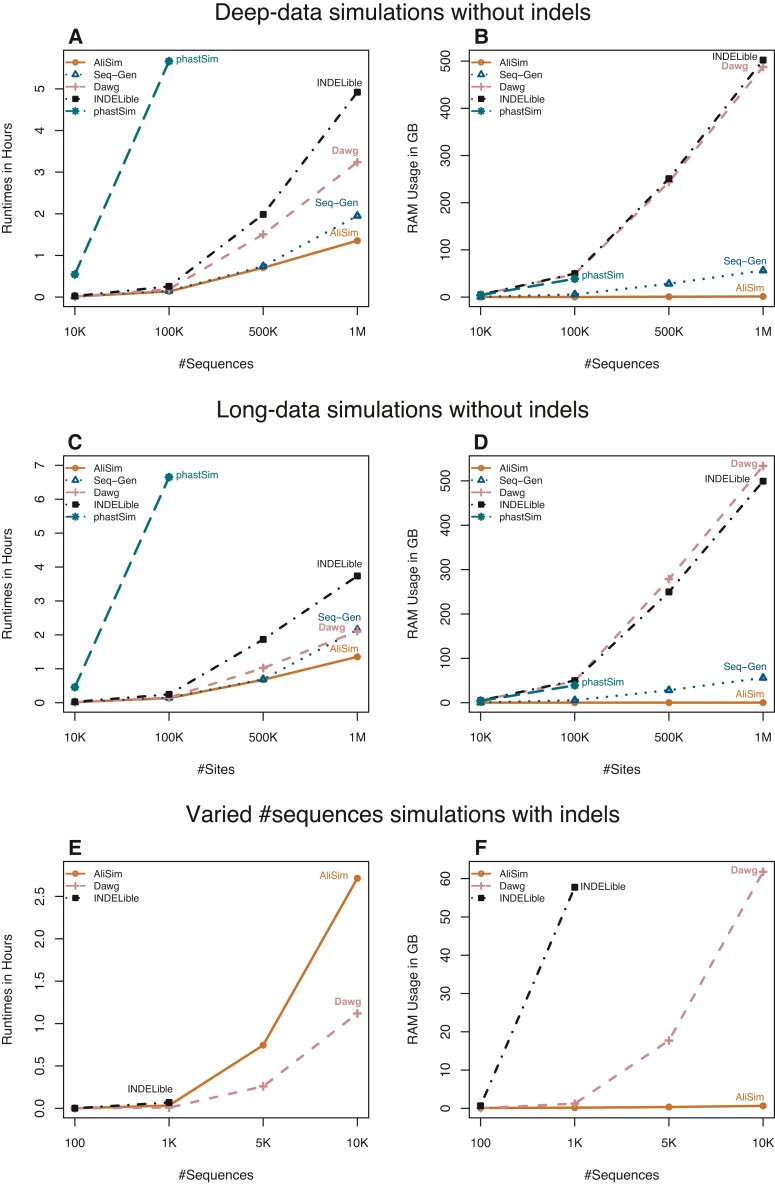
Similar articles
-
AliSim-HPC: parallel sequence simulator for phylogenetics.Bioinformatics. 2023 Sep 2;39(9):btad540. doi: 10.1093/bioinformatics/btad540. Bioinformatics. 2023. PMID: 37656933 Free PMC article.
-
IQ-TREE 2: New Models and Efficient Methods for Phylogenetic Inference in the Genomic Era.Mol Biol Evol. 2020 May 1;37(5):1530-1534. doi: 10.1093/molbev/msaa015. Mol Biol Evol. 2020. PMID: 32011700 Free PMC article.
-
DNA assembly with gaps (Dawg): simulating sequence evolution.Bioinformatics. 2005 Nov 1;21 Suppl 3:iii31-8. doi: 10.1093/bioinformatics/bti1200. Bioinformatics. 2005. PMID: 16306390
-
Molecular Phylogenetics: Concepts for a Newcomer.Adv Biochem Eng Biotechnol. 2017;160:185-196. doi: 10.1007/10_2016_49. Adv Biochem Eng Biotechnol. 2017. PMID: 27783136 Review.
-
Phylogenetics of modern birds in the era of genomics.Proc Biol Sci. 2005 May 22;272(1567):979-92. doi: 10.1098/rspb.2004.3035. Proc Biol Sci. 2005. PMID: 16024355 Free PMC article. Review.
Cited by
-
Scoutknife: A naïve, whole genome informed phylogenetic robusticity metric.F1000Res. 2024 Jul 10;12:945. doi: 10.12688/f1000research.139356.2. eCollection 2023. F1000Res. 2024. PMID: 38799242 Free PMC article.
-
Insertions and Deletions: Computational Methods, Evolutionary Dynamics, and Biological Applications.Mol Biol Evol. 2024 Sep 4;41(9):msae177. doi: 10.1093/molbev/msae177. Mol Biol Evol. 2024. PMID: 39172750 Free PMC article. Review.
-
MAST: Phylogenetic Inference with Mixtures Across Sites and Trees.Syst Biol. 2024 Jul 27;73(2):375-391. doi: 10.1093/sysbio/syae008. Syst Biol. 2024. PMID: 38421146 Free PMC article.
-
Reliable estimation of tree branch lengths using deep neural networks.PLoS Comput Biol. 2024 Aug 5;20(8):e1012337. doi: 10.1371/journal.pcbi.1012337. eCollection 2024 Aug. PLoS Comput Biol. 2024. PMID: 39102450 Free PMC article.
-
nT4X and nT4M: Novel Time Non-reversible Mixture Amino Acid Substitution Models.J Mol Evol. 2025 Feb;93(1):136-148. doi: 10.1007/s00239-024-10230-8. Epub 2025 Jan 20. J Mol Evol. 2025. PMID: 39832000
References
-
- Abadi S, Avram O, Rosset S, Pupko T, Mayrose I. 2020. ModelTeller: model selection for optimal phylogenetic reconstruction using machine learning. Mol Biol Evol. 37(11):3338–3352. - PubMed
-
- Adell JC, Dopazo J. 1994. Monte Carlo simulation in phylogenies: an application to test the constancy of evolutionary rates. J Mol Evol. 38(3):305–309. - PubMed
-
- Benner SA, Cohen MA, Gonnet GH. 1993. Empirical and structural models for insertions and deletions in the divergent evolution of proteins. J Mol Biol. 229(4):1065–1082. - PubMed
-
- Cartwright RA. 2005. DNA assembly with gaps (Dawg): simulating sequence evolution. Bioinformatics 21(Suppl. 3):31–38. - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
