

Loss of mitochondrial ATPase ATAD3A contributes to nonalcoholic fatty liver disease through accumulation of lipids and damaged mitochondria
- PMID: 35513069
- PMCID: PMC9157002
- DOI: 10.1016/j.jbc.2022.102008
Loss of mitochondrial ATPase ATAD3A contributes to nonalcoholic fatty liver disease through accumulation of lipids and damaged mitochondria
Abstract
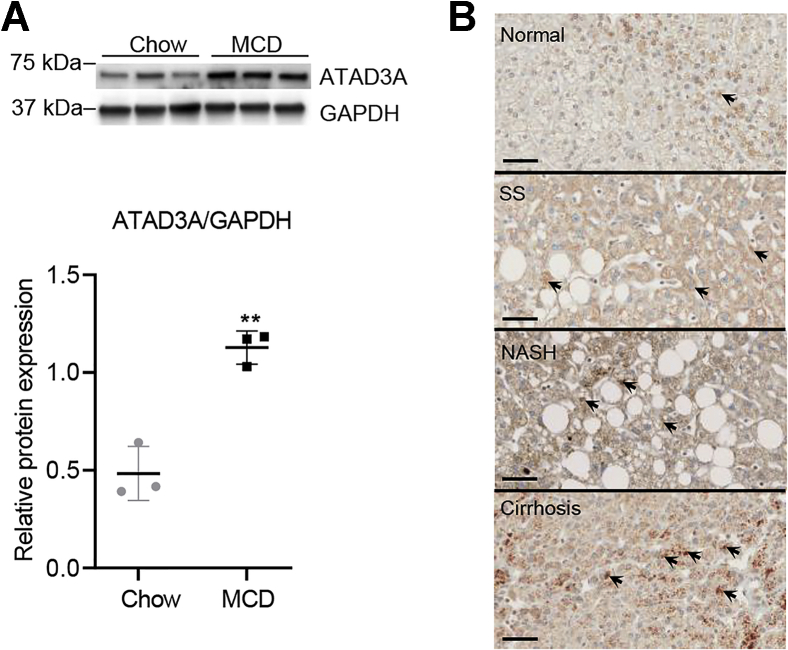
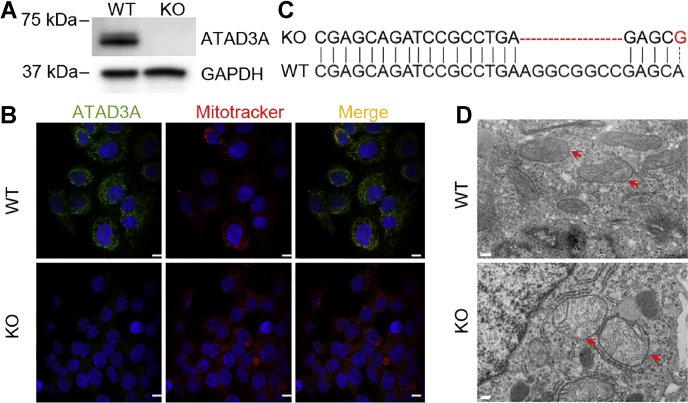
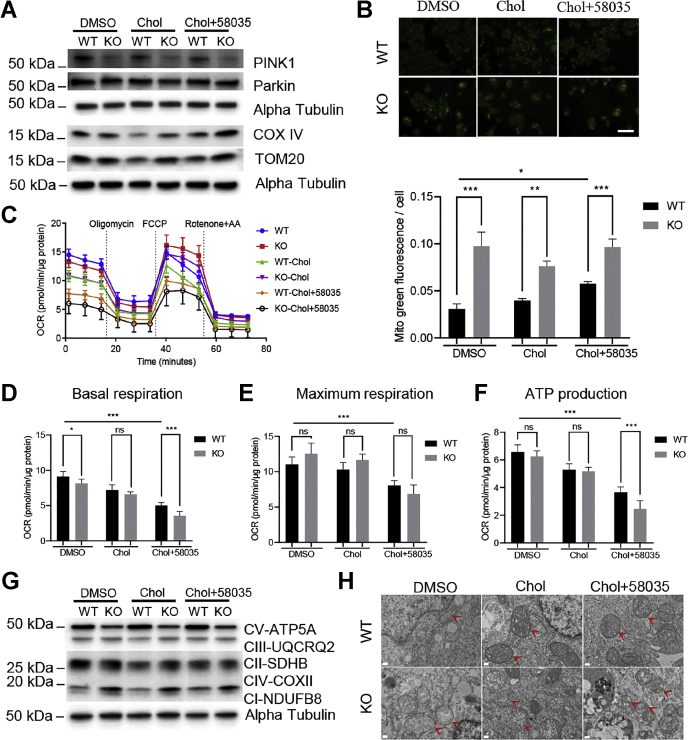
Mitochondrial ATPase ATAD3A is essential for cholesterol transport, mitochondrial structure, and cell survival. However, the relationship between ATAD3A and nonalcoholic fatty liver disease (NAFLD) is largely unknown. In this study, we found that ATAD3A was upregulated in the progression of NAFLD in livers from rats with diet-induced nonalcoholic steatohepatitis and in human livers from patients diagnosed with NAFLD. We used CRISPR-Cas9 to delete ATAD3A in Huh7 human hepatocellular carcinoma cells and used RNAi to silence ATAD3A expression in human hepatocytes isolated from humanized liver-chimeric mice to assess the influence of ATAD3A deletion on liver cells with free cholesterol (FC) overload induced by treatment with cholesterol plus 58035, an inhibitor of acetyl-CoA acetyltransferase. Our results showed that ATAD3A KO exacerbated FC accumulation under FC overload in Huh7 cells and also that triglyceride levels were significantly increased in ATAD3A KO Huh7 cells following inhibition of lipolysis mediated by upregulation of lipid droplet-binding protein perilipin-2. Moreover, loss of ATAD3A upregulated autophagosome-associated light chain 3-II protein and p62 in Huh7 cells and fresh human hepatocytes through blockage of autophagosome degradation. Finally, we show the mitophagy mediator, PTEN-induced kinase 1, was downregulated in ATAD3A KO Huh7 cells, suggesting that ATAD3A KO inhibits mitophagy. These results also showed that loss of ATAD3A impaired mitochondrial basal respiration and ATP production in Huh7 cells under FC overload, accompanied by downregulation of mitochondrial ATP synthase. Taken together, we conclude that loss of ATAD3A promotes the progression of NAFLD through the accumulation of FC, triglyceride, and damaged mitochondria in hepatocytes.
Keywords: ATAD3A; NAFLD; autophagy; cholesterol; fatty acid oxidation; free fatty acid; mitochondrial respiration; mitophagy; triglyceride.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Conflict of interest The authors declare that they have no conflicts of interest with the contents of this article.
Figures




References
-
- Younossi Z., Anstee Q.M., Marietti M., Hardy T., Henry L., Eslam M., et al. Global burden of NAFLD and NASH: trends, predictions, risk factors and prevention. Nat. Rev. Gastroenterol. Hepatol. 2018;15:11–20. - PubMed
-
- Huang T.D., Behary J., Zekry A. Non-alcoholic fatty liver disease: a review of epidemiology, risk factors, diagnosis and management. Intern. Med. J. 2020;50:1038–1047. - PubMed
-
- Arguello G., Balboa E., Arrese M., Zanlungo S. Recent insights on the role of cholesterol in non-alcoholic fatty liver disease. Biochim. Biophys. Acta. 2015;1852:1765–1778. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Research Materials
