

SPIN enables high throughput species identification of archaeological bone by proteomics
- PMID: 35513387
- PMCID: PMC9072323
- DOI: 10.1038/s41467-022-30097-x
SPIN enables high throughput species identification of archaeological bone by proteomics
Abstract
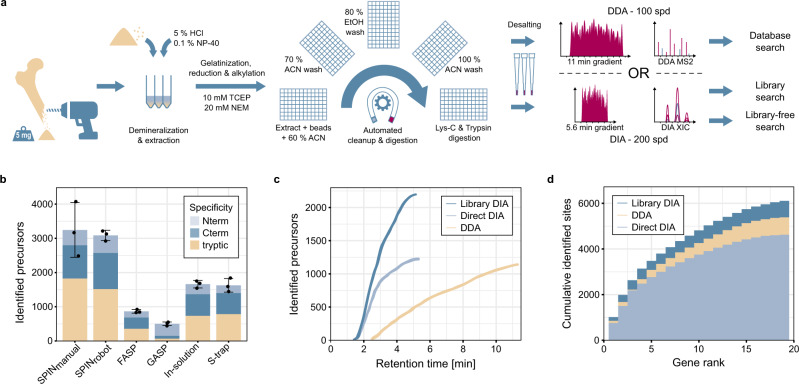
Species determination based on genetic evidence is an indispensable tool in archaeology, forensics, ecology, and food authentication. Most available analytical approaches involve compromises with regard to the number of detectable species, high cost due to low throughput, or a labor-intensive manual process. Here, we introduce "Species by Proteome INvestigation" (SPIN), a shotgun proteomics workflow for analyzing archaeological bone capable of querying over 150 mammalian species by liquid chromatography-tandem mass spectrometry (LC-MS/MS). Rapid peptide chromatography and data-independent acquisition (DIA) with throughput of 200 samples per day reduce expensive MS time, whereas streamlined sample preparation and automated data interpretation save labor costs. We confirm the successful classification of known reference bones, including domestic species and great apes, beyond the taxonomic resolution of the conventional peptide mass fingerprinting (PMF)-based Zooarchaeology by Mass Spectrometry (ZooMS) method. In a blinded study of degraded Iron-Age material from Scandinavia, SPIN produces reproducible results between replicates, which are consistent with morphological analysis. Finally, we demonstrate the high throughput capabilities of the method in a high-degradation context by analyzing more than two hundred Middle and Upper Palaeolithic bones from Southern European sites with late Neanderthal occupation. While this initial study is focused on modern and archaeological mammalian bone, SPIN will be open and expandable to other biological tissues and taxa.
© 2022. The Author(s).
Conflict of interest statement
The authors declare no competing interests.
Figures





References
-
- Asensio L, González I, García T, Martín R. Determination of food authenticity by enzyme-linked immunosorbent assay (ELISA) Food Control. 2008;19:1–8. doi: 10.1016/j.foodcont.2007.02.010. - DOI
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Molecular Biology Databases
