

Distinct Patterns of Clonal Evolution Drive Myelodysplastic Syndrome Progression to Secondary Acute Myeloid Leukemia
- PMID: 35522837
- PMCID: PMC9610896
- DOI: 10.1158/2643-3230.BCD-21-0128
Distinct Patterns of Clonal Evolution Drive Myelodysplastic Syndrome Progression to Secondary Acute Myeloid Leukemia
Abstract
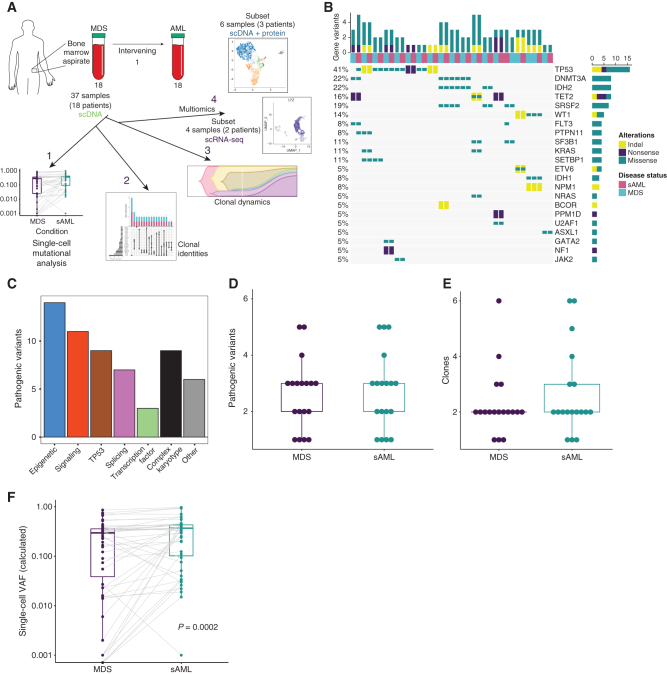
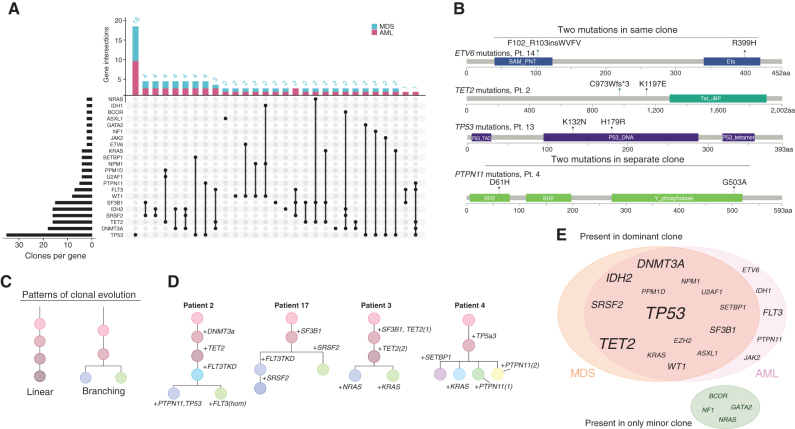
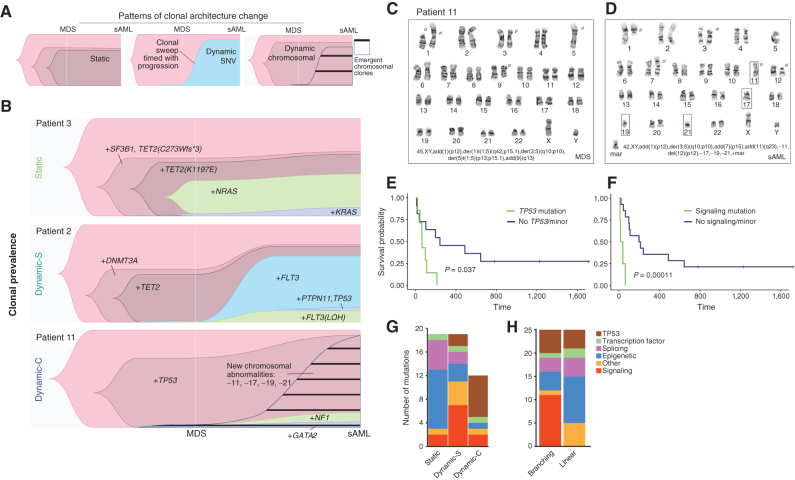
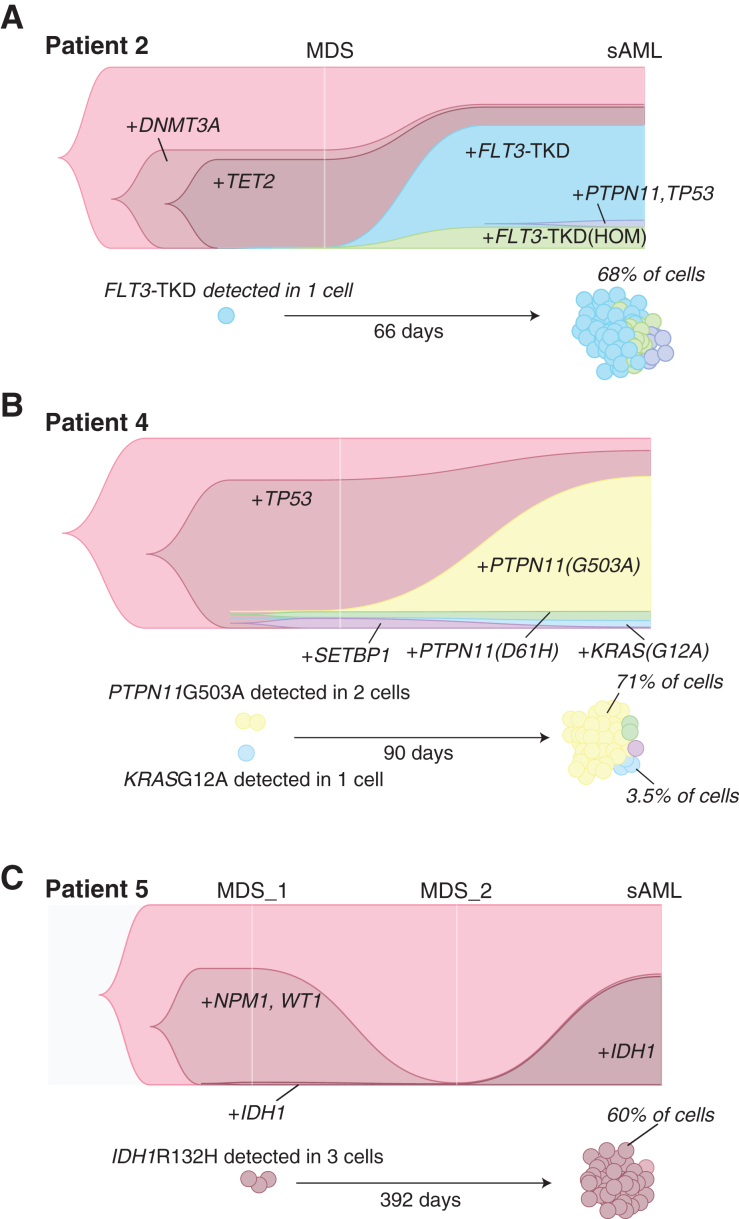
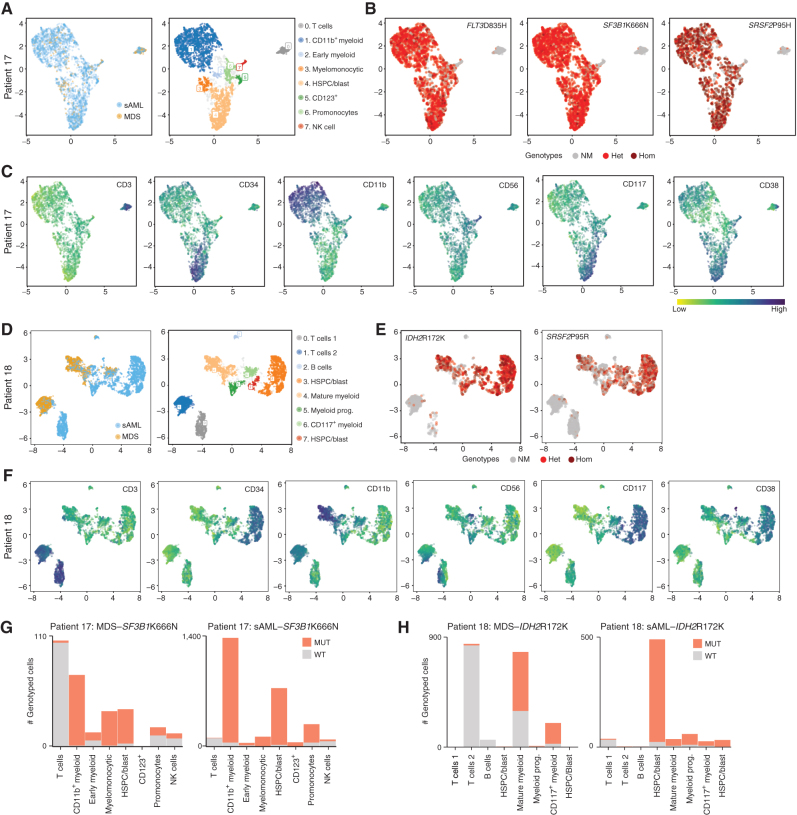
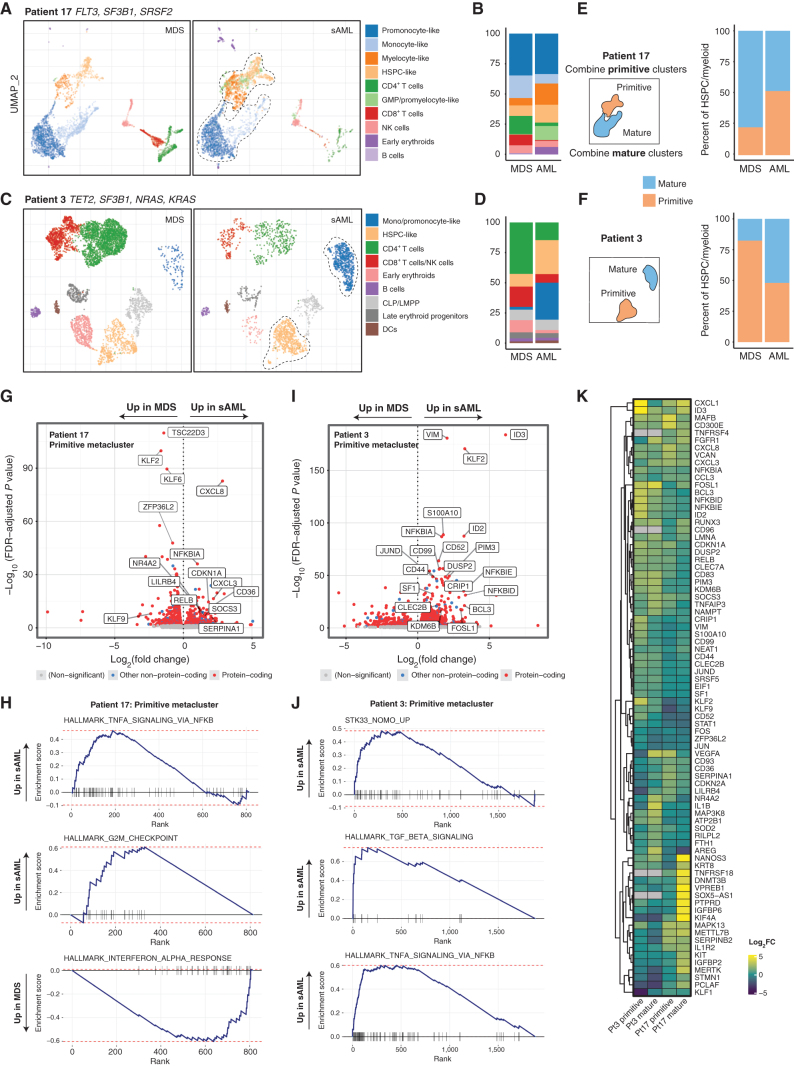
Clonal evolution in myelodysplastic syndrome (MDS) can result in clinical progression and secondary acute myeloid leukemia (sAML). To dissect changes in clonal architecture associated with this progression, we performed single-cell genotyping of paired MDS and sAML samples from 18 patients. Analysis of single-cell genotypes revealed patient-specific clonal evolution and enabled the assessment of single-cell mutational cooccurrence. We discovered that changes in clonal architecture proceed via distinct patterns, classified as static or dynamic, with dynamic clonal architectures having a more proliferative phenotype by blast count fold change. Proteogenomic analysis of a subset of patients confirmed that pathogenic mutations were primarily confined to primitive and mature myeloid cells, though we also identify rare but present mutations in lymphocyte subsets. Single-cell transcriptomic analysis of paired sample sets further identified gene sets and signaling pathways involved in two cases of progression. Together, these data define serial changes in the MDS clonal landscape with clinical and therapeutic implications.
Significance: Precise clonal trajectories in MDS progression are made possible by single-cell genomic sequencing. Here we use this technology to uncover the patterns of clonal architecture and clonal evolution that drive the transformation to secondary AML. We further define the phenotypic and transcriptional changes of disease progression at the single-cell level. See related article by Menssen et al., p. 330 (31). See related commentary by Romine and van Galen, p. 270. This article is highlighted in the In This Issue feature, p. 265.
©2022 American Association for Cancer Research.
Figures
Comment in
-
Rise of the Clones: Myelodysplastic Syndrome to Secondary Acute Myeloid Leukemia.Blood Cancer Discov. 2022 Jul 6;3(4):270-272. doi: 10.1158/2643-3230.BCD-22-0046. Blood Cancer Discov. 2022. PMID: 35709709 Free PMC article.
References
-
- Kuykendall A, Duployez N, Boissel N, Lancet JE, Welch JS. Acute myeloid leukemia: the good, the bad, and the ugly. Am Soc Clin Oncol Educ Book 2018;38:555–73. - PubMed
-
- Bennett JM. Secondary acute myeloid leukemia. Leuk Res 1995;19:231–2. - PubMed
-
- Weinberg OK, Seetharam M, Ren L, Seo K, Ma L, Merker JD, et al. Clinical characterization of acute myeloid leukemia with myelodysplasia-related changes as defined by the 2008 WHO classification system. Blood 2009;113:1906–8. - PubMed
-
- Nowell PC. The clonal evolution of tumor cell populations. Science 1976;194:23–8. - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
