

HSP90 N-terminal inhibitors target oncoprotein MORC2 for autophagic degradation and suppress MORC2-driven breast cancer progression
- PMID: 35522895
- PMCID: PMC9076019
- DOI: 10.1002/ctm2.825
HSP90 N-terminal inhibitors target oncoprotein MORC2 for autophagic degradation and suppress MORC2-driven breast cancer progression
Abstract
Aims: MORC family CW-type zinc finger 2 (MORC2), a GHKL-type ATPase, is aberrantly upregulated in multiple types of human tumors with profound effects on cancer aggressiveness, therapeutic resistance, and clinical outcome, thus making it an attractive drug target for anticancer therapy. However, the antagonists of MORC2 have not yet been documented.
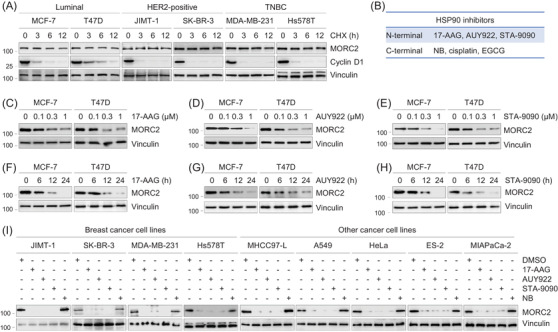
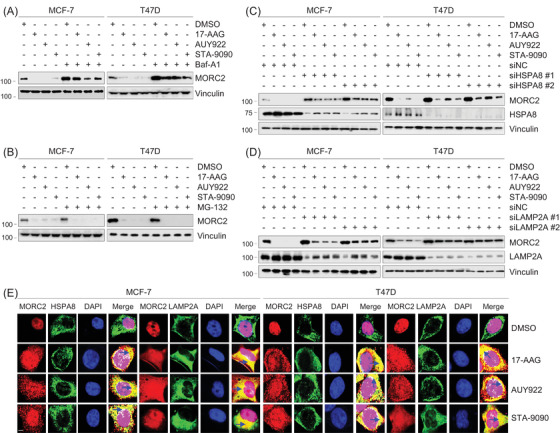
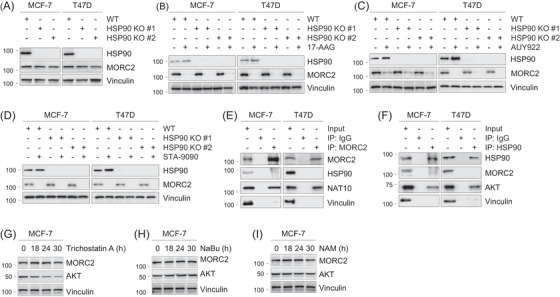
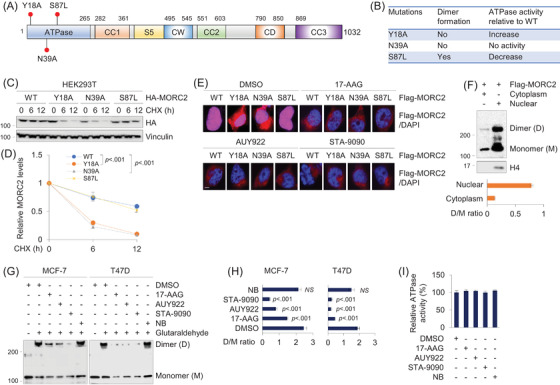
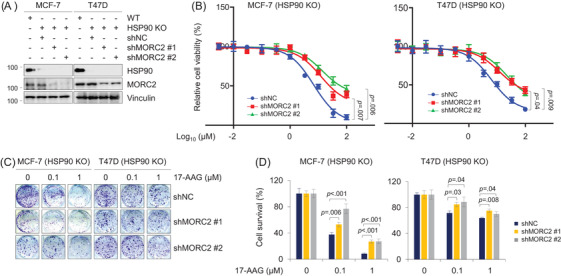
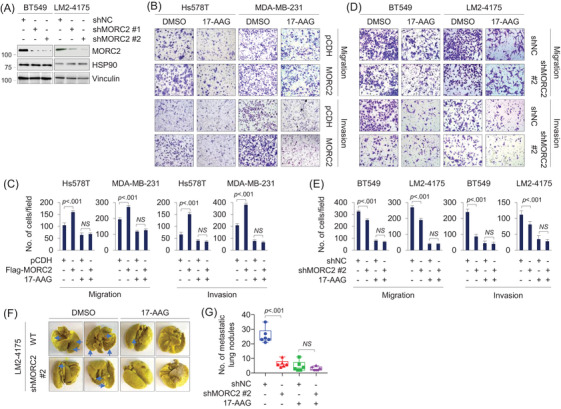
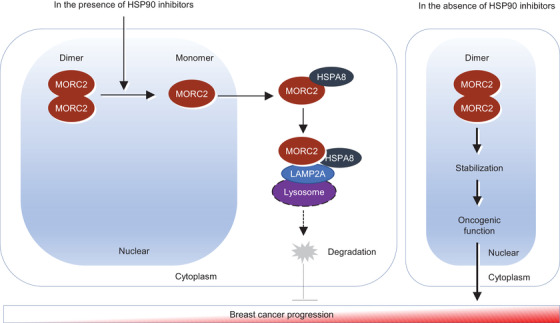
Methods and results: We report that MORC2 is a relatively stable protein, and the N-terminal homodimerization but not ATP binding and hydrolysis is crucial for its stability through immunoblotting analysis and Quantitative real-time PCR. The N-terminal but not C-terminal inhibitors of heat shock protein 90 (HSP90) destabilize MORC2 in multiple cancer cell lines, and strikingly, this process is independent on HSP90. Mechanistical investigations revealed that HSP90 N-terminal inhibitors disrupt MORC2 homodimer formation without affecting its ATPase activities, and promote its lysosomal degradation through the chaperone-mediated autophagy pathway. Consequently, HSP90 inhibitor 17-AAG effectively blocks the growth and metastatic potential of MORC2-expressing breast cancer cells both in vitro and in vivo, and these noted effects are not due to HSP90 inhibition.
Conclusion: We uncover a previously unknown role for HSP90 N-terminal inhibitors in promoting MORC2 degradation in a HSP90-indepentent manner and support the potential application of these inhibitors for treating MORC2-overexpressing tumors, even those with low or absent HSP90 expression. These results also provide new clue for further design of novel small-molecule inhibitors of MORC2 for anticancer therapeutic application.
Keywords: HSP90 inhibitor; MORC2; breast cancer; chaperone-mediated autophagy; protein degradation.
© 2022 The Authors. Clinical and Translational Medicine published by John Wiley & Sons Australia, Ltd on behalf of Shanghai Institute of Clinical Bioinformatics.
Conflict of interest statement
The authors declare that they have no conflict of interest.
Figures
Similar articles
-
Stabilization of MORC2 by estrogen and antiestrogens through GPER1- PRKACA-CMA pathway contributes to estrogen-induced proliferation and endocrine resistance of breast cancer cells.Autophagy. 2020 Jun;16(6):1061-1076. doi: 10.1080/15548627.2019.1659609. Epub 2019 Sep 6. Autophagy. 2020. PMID: 32401166 Free PMC article.
-
Destabilization of microrchidia family CW-type zinc finger 2 via the cyclin-dependent kinase 1-chaperone-mediated autophagy pathway promotes mitotic arrest and enhances cancer cellular sensitivity to microtubule-targeting agents.Clin Transl Med. 2023 Mar;13(3):e1210. doi: 10.1002/ctm2.1210. Clin Transl Med. 2023. PMID: 36967563 Free PMC article.
-
Dynamic SUMOylation of MORC2 orchestrates chromatin remodelling and DNA repair in response to DNA damage and drives chemoresistance in breast cancer.Theranostics. 2023 Jan 22;13(3):973-990. doi: 10.7150/thno.79688. eCollection 2023. Theranostics. 2023. PMID: 36793866 Free PMC article.
-
Anticancer Inhibitors of Hsp90 Function: Beyond the Usual Suspects.Adv Cancer Res. 2016;129:51-88. doi: 10.1016/bs.acr.2015.12.001. Epub 2016 Feb 10. Adv Cancer Res. 2016. PMID: 26916001 Free PMC article. Review.
-
Hsp90: an emerging target for breast cancer therapy.Anticancer Drugs. 2004 Aug;15(7):651-62. doi: 10.1097/01.cad.0000136876.11928.be. Anticancer Drugs. 2004. PMID: 15269596 Review.
Cited by
-
MORC2 regulates RBM39-mediated CDK5RAP2 alternative splicing to promote EMT and metastasis in colon cancer.Cell Death Dis. 2024 Jul 24;15(7):530. doi: 10.1038/s41419-024-06908-y. Cell Death Dis. 2024. PMID: 39048555 Free PMC article.
-
Emerging roles of the chromatin remodeler MORC2 in cancer metabolism.Med Oncol. 2024 Aug 8;41(9):221. doi: 10.1007/s12032-024-02464-9. Med Oncol. 2024. PMID: 39117768 Review.
-
Novel Insights into the Role of Chromatin Remodeler MORC2 in Cancer.Biomolecules. 2023 Oct 15;13(10):1527. doi: 10.3390/biom13101527. Biomolecules. 2023. PMID: 37892209 Free PMC article. Review.
-
Heat Shock Proteins and Breast Cancer.Int J Mol Sci. 2024 Jan 10;25(2):876. doi: 10.3390/ijms25020876. Int J Mol Sci. 2024. PMID: 38255948 Free PMC article. Review.
-
Oncogenic MORC2 in cancer development and beyond.Genes Dis. 2023 Jul 3;11(2):861-873. doi: 10.1016/j.gendis.2023.05.010. eCollection 2024 Mar. Genes Dis. 2023. PMID: 37692502 Free PMC article. Review.
References
Publication types
MeSH terms
Substances
LinkOut - more resources
Full Text Sources
Medical
Research Materials