

Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution
- PMID: 35523183
- PMCID: PMC9452598
- DOI: 10.1016/j.cell.2022.04.015
Lineage tracing reveals the phylodynamics, plasticity, and paths of tumor evolution
Abstract
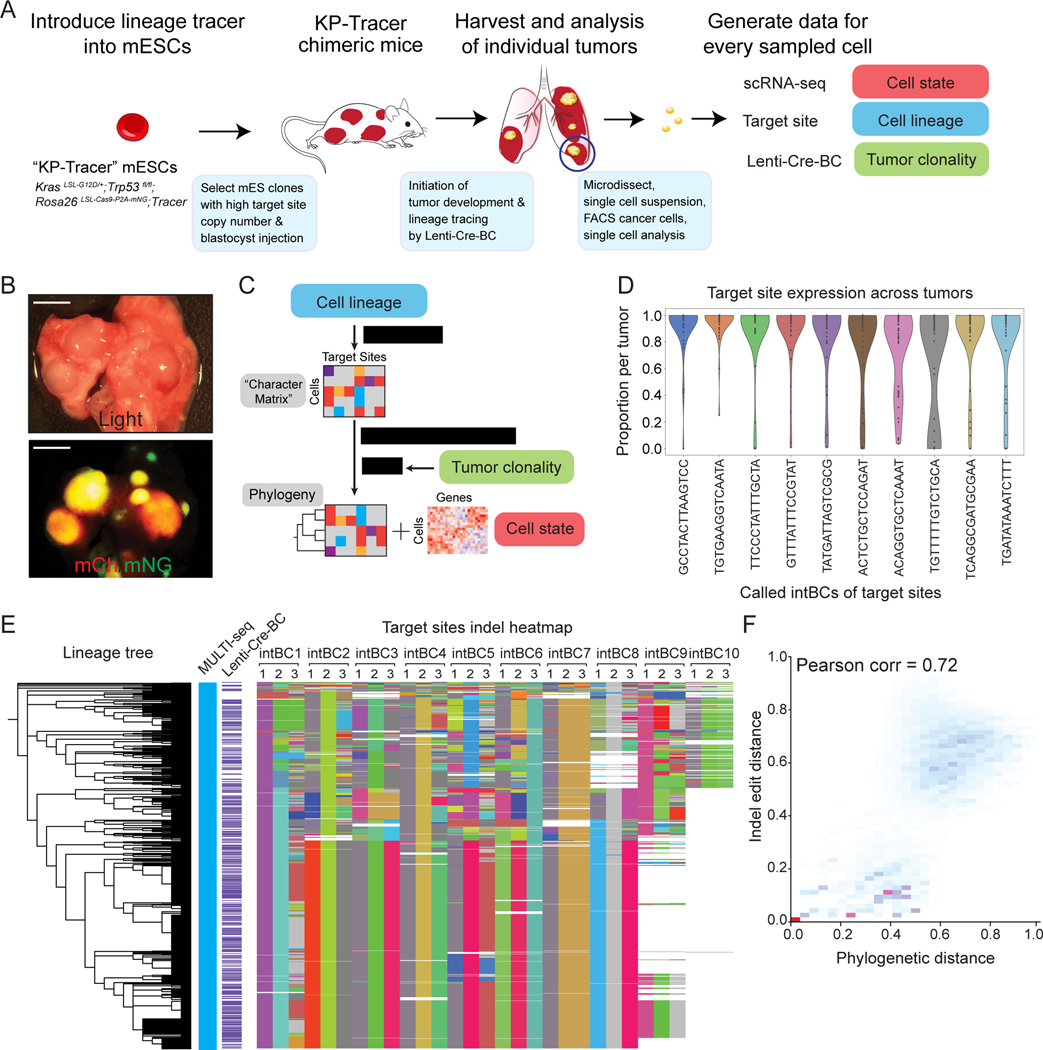
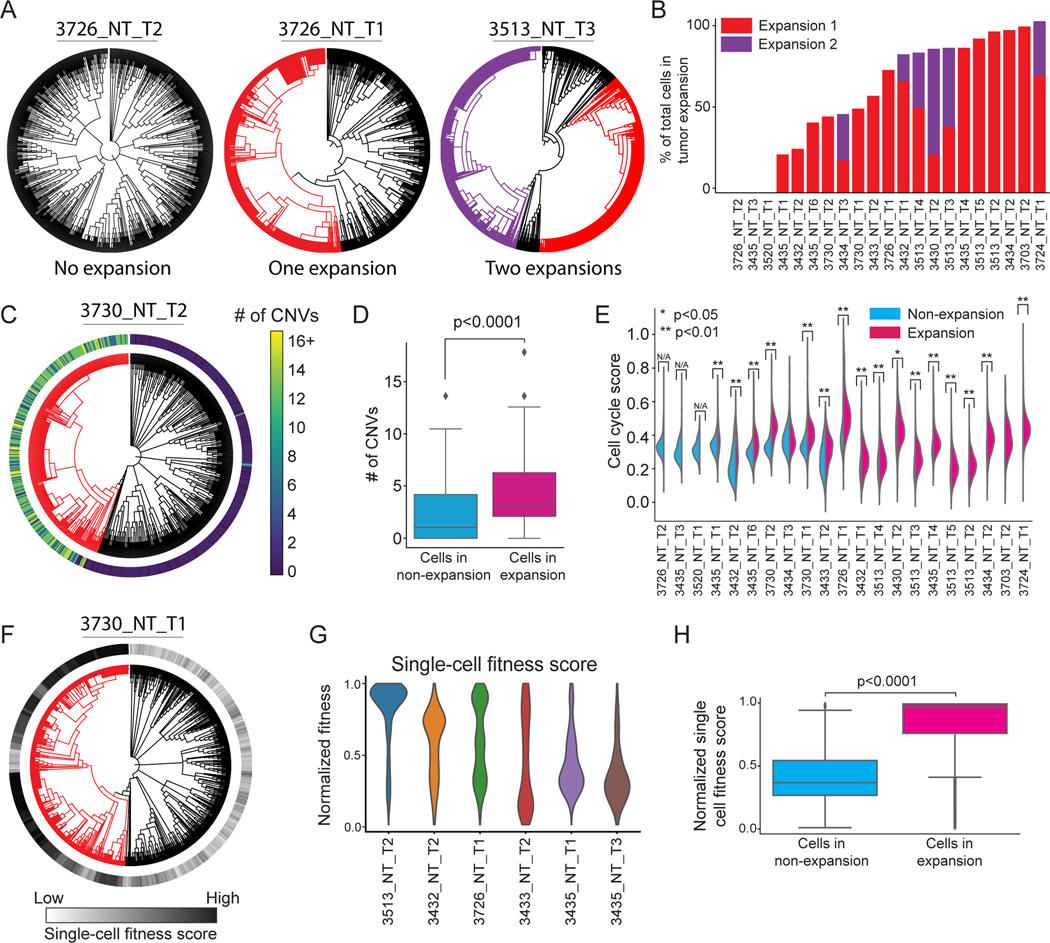
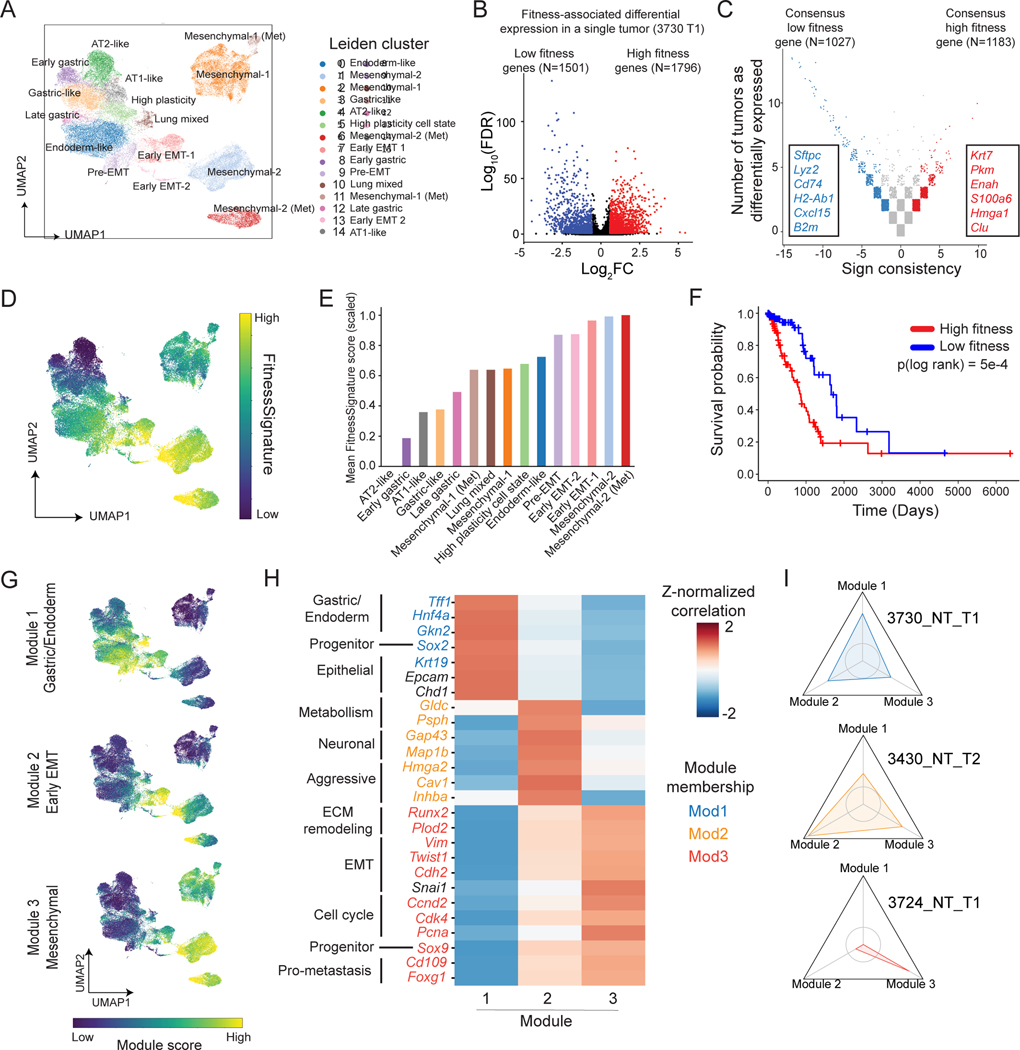
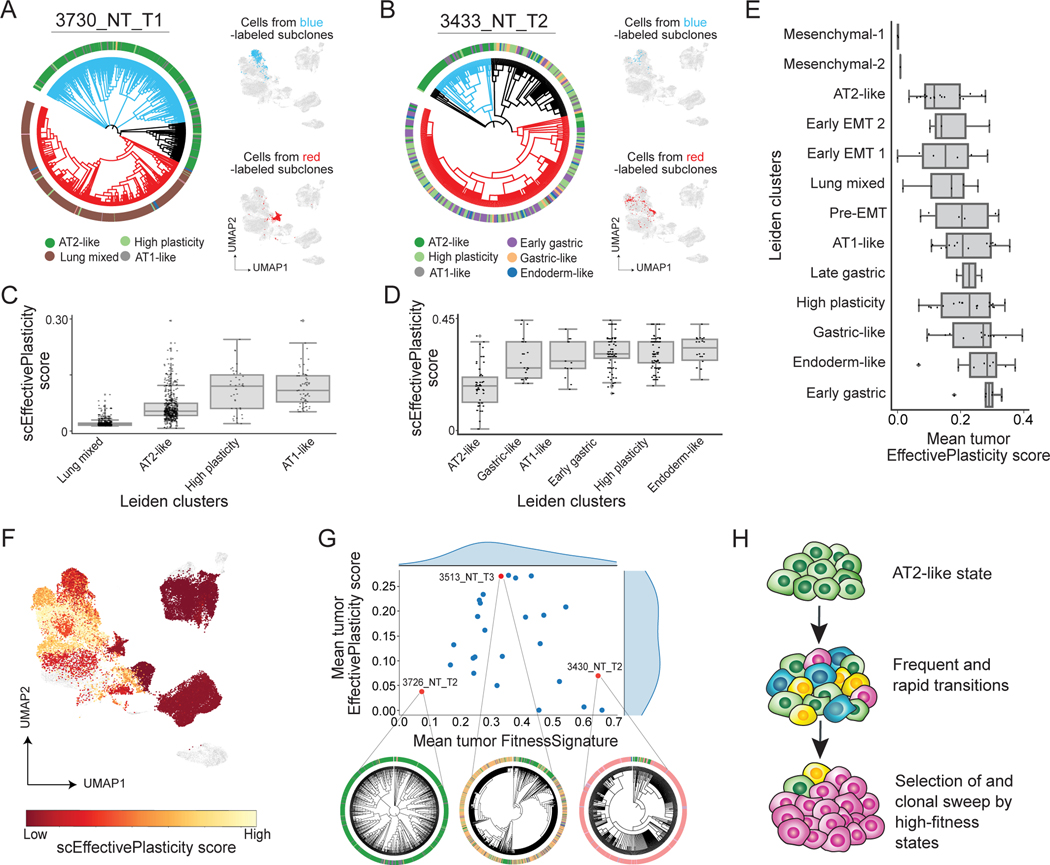
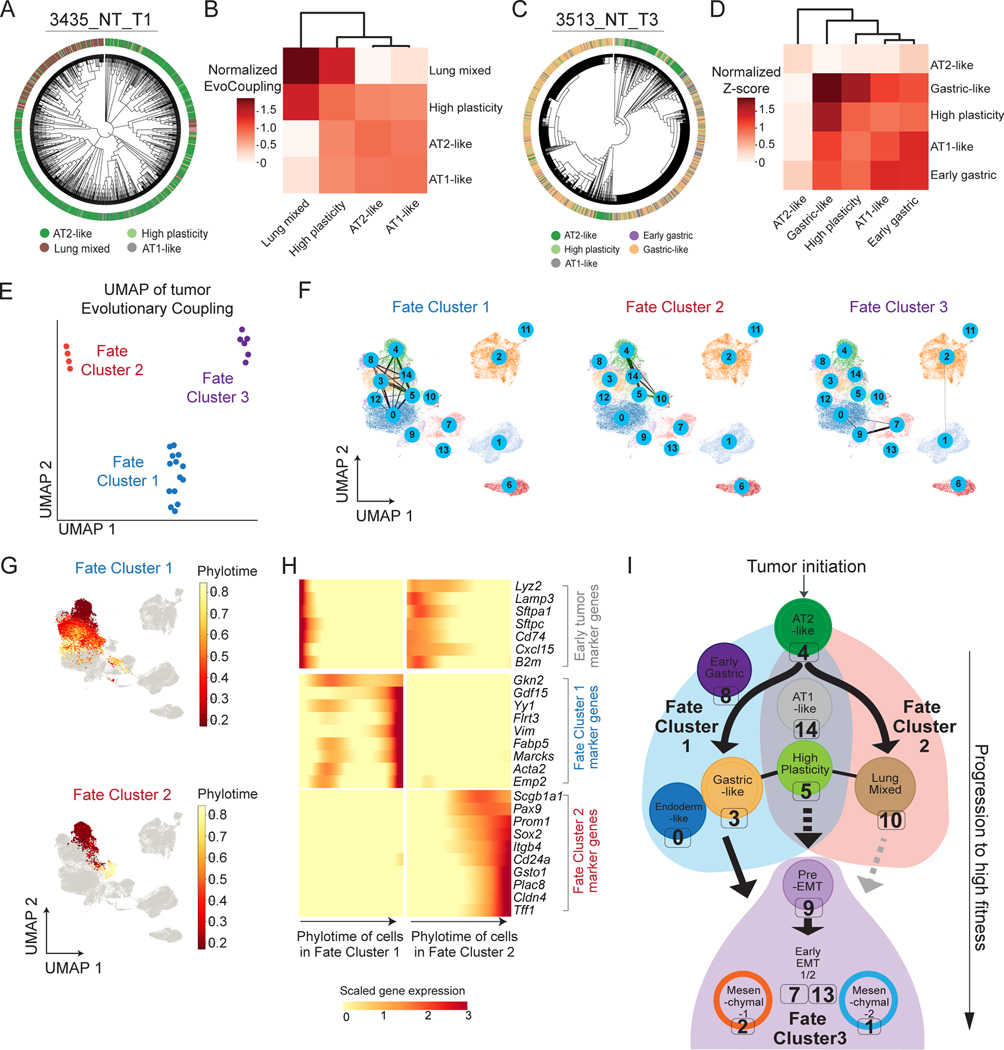
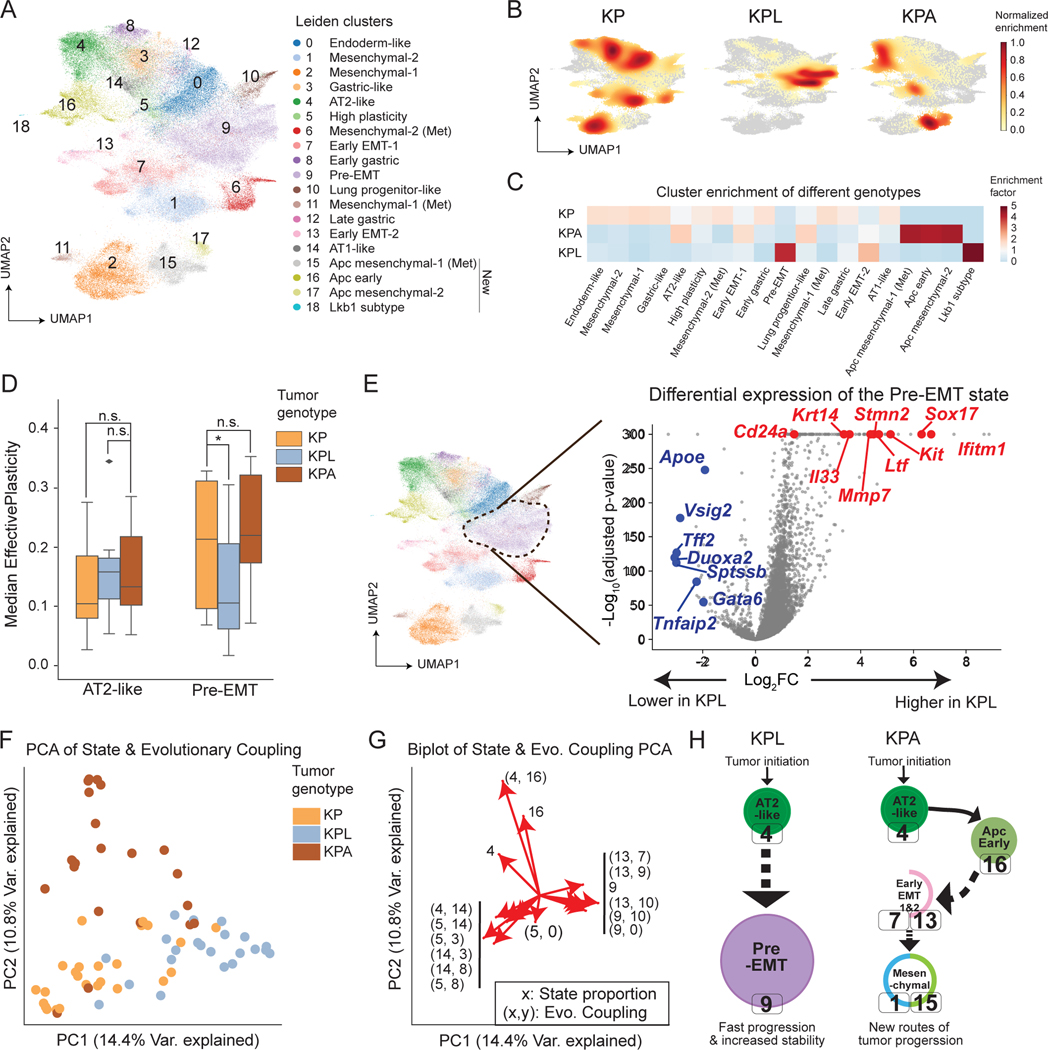
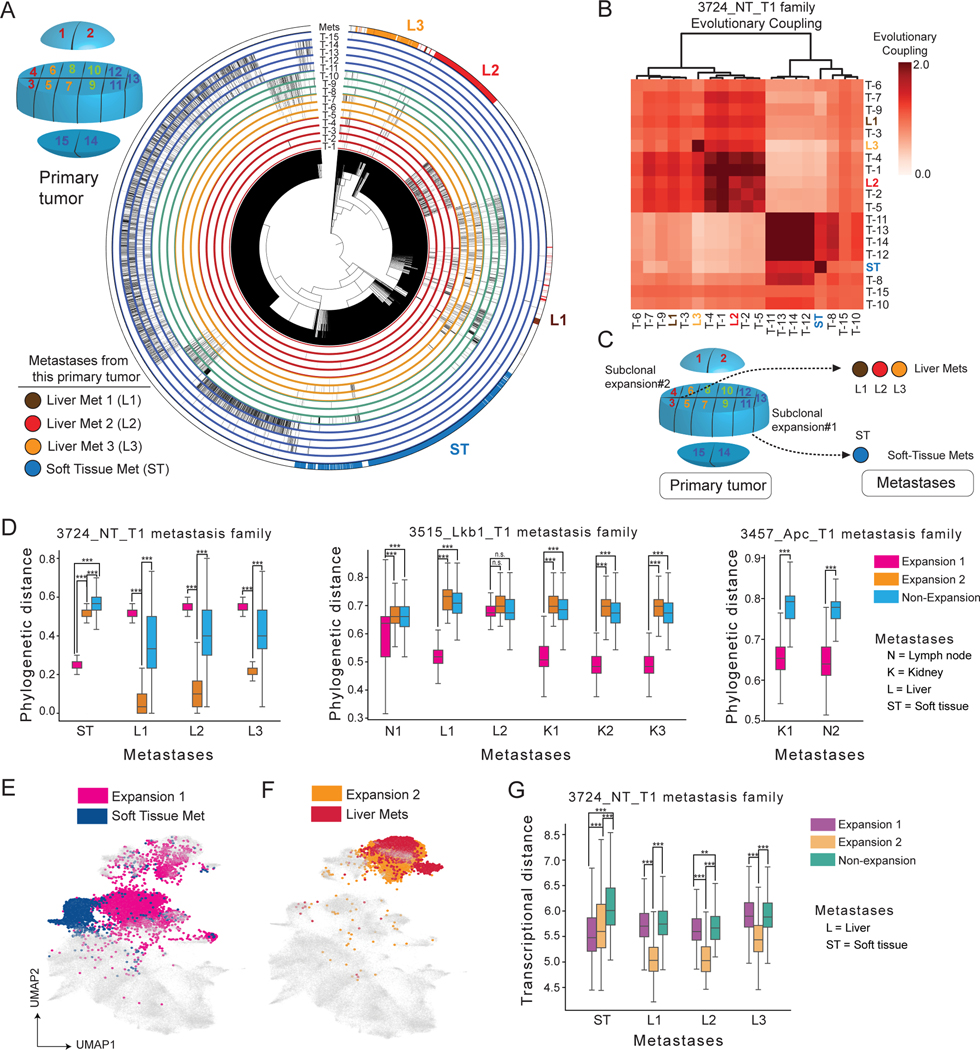
Tumor evolution is driven by the progressive acquisition of genetic and epigenetic alterations that enable uncontrolled growth and expansion to neighboring and distal tissues. The study of phylogenetic relationships between cancer cells provides key insights into these processes. Here, we introduced an evolving lineage-tracing system with a single-cell RNA-seq readout into a mouse model of Kras;Trp53(KP)-driven lung adenocarcinoma and tracked tumor evolution from single-transformed cells to metastatic tumors at unprecedented resolution. We found that the loss of the initial, stable alveolar-type2-like state was accompanied by a transient increase in plasticity. This was followed by the adoption of distinct transcriptional programs that enable rapid expansion and, ultimately, clonal sweep of stable subclones capable of metastasizing. Finally, tumors develop through stereotypical evolutionary trajectories, and perturbing additional tumor suppressors accelerates progression by creating novel trajectories. Our study elucidates the hierarchical nature of tumor evolution and, more broadly, enables in-depth studies of tumor progression.
Keywords: fitness; genetically engineered mouse model; lineage tracing; lung cancer; phylogenetics; plasticity; single cell; transcriptome heterogeneity; tumor evolution.
Copyright © 2022 The Authors. Published by Elsevier Inc. All rights reserved.
Conflict of interest statement
Declaration of interests J.S.W. declares outside interest in 5 AM Venture, Amgen, Chroma Medicine, KSQ Therapeutics, Maze Therapeutics, Tenaya Therapeutics, and Tessera Therapeutics. T.J. is a member of the Board of Directors of Amgen and Thermo Fisher Scientific, is a co-founder of Dragonfly Therapeutics and T2 Biosystems, and is the president of Break Through Cancer. T.J. serves on the Scientific Advisory Board of Dragonfly Therapeutics, SQZ Biotech, and Skyhawk Therapeutics. None of these affiliations represent a conflict of interest with respect to this study. T.G.B. is an advisor to Array BioPharma, Revolution Medicines, Novartis, AstraZeneca, Takeda, Springworks, Jazz Pharmaceuticals, Relay Therapeutics, Rain Therapeutics, and Engine Biosciences and receives research funding from Novartis, Strategia, Kinnate, and Revolution Medicines. J.M.R. consults for Maze Therapeutics and Waypoint Bio. Z.J.G. is an equity holder in Scribe Biosciences and Provenance bio and a member of the SAB of Serotiny Bio.
Figures
Comment in
-
"Fateful" encounter: Lineage tracing meets phylogeny to unravel mysteries of cancer progression.Dev Cell. 2022 Jul 25;57(14):1680-1682. doi: 10.1016/j.devcel.2022.07.002. Dev Cell. 2022. PMID: 35901781
References
-
- Alemany Anna, Florescu Maria, Baron Chloé S., Peterson-Maduro Josi, and van Oudenaarden Alexander. 2018. “Whole-Organism Clone Tracing Using Single-Cell Sequencing.” Nature 556 (7699): 108–12. - PubMed
-
- Amirouchene-Angelozzi Nabil, Swanton Charles, and Bardelli Alberto. 2017. “Tumor Evolution as a Therapeutic Target.” Cancer Discovery 7(8), pp.805–817. - PubMed
Publication types
MeSH terms
Grants and funding
- T32 GM007287/GM/NIGMS NIH HHS/United States
- U01 CA217882/CA/NCI NIH HHS/United States
- P30 CA014051/CA/NCI NIH HHS/United States
- T32 GM007753/GM/NIGMS NIH HHS/United States
- F31 CA257349/CA/NCI NIH HHS/United States
- U01 CA272546/CA/NCI NIH HHS/United States
- F32 GM125247/GM/NIGMS NIH HHS/United States
- R01 CA204302/CA/NCI NIH HHS/United States
- F31 NS115380/NS/NINDS NIH HHS/United States
- U54 CA224081/CA/NCI NIH HHS/United States
- RM1 HG009490/HG/NHGRI NIH HHS/United States
- HHMI/Howard Hughes Medical Institute/United States
- R01 CA211052/CA/NCI NIH HHS/United States
- R01 CA231300/CA/NCI NIH HHS/United States
- R01 CA169338/CA/NCI NIH HHS/United States
- F32 GM128366/GM/NIGMS NIH HHS/United States
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
Research Materials
Miscellaneous
