

Evaluating metagenomic assembly approaches for biome-specific gene catalogues
- PMID: 35524337
- PMCID: PMC9074274
- DOI: 10.1186/s40168-022-01259-2
Evaluating metagenomic assembly approaches for biome-specific gene catalogues
Abstract
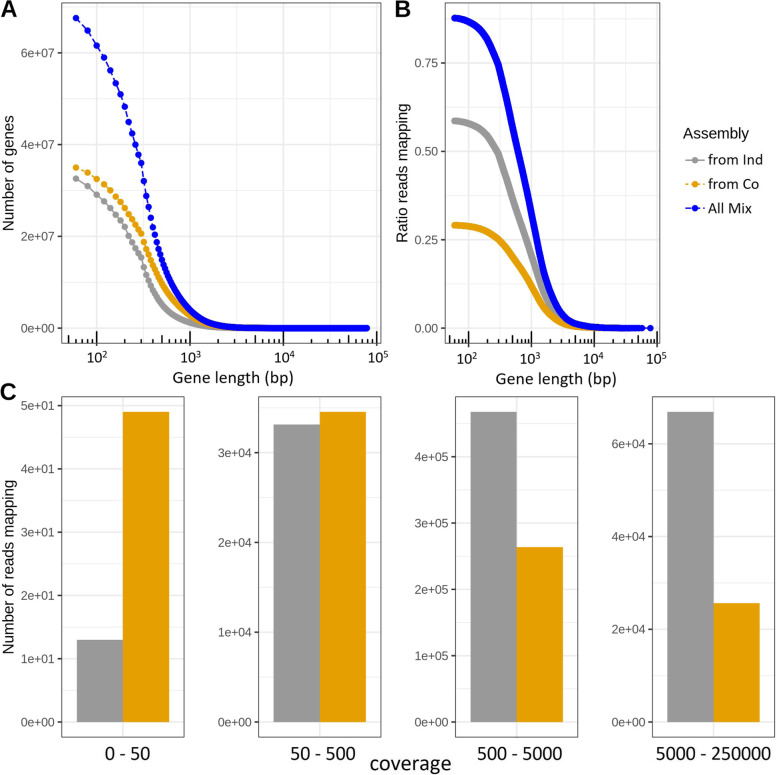
Background: For many environments, biome-specific microbial gene catalogues are being recovered using shotgun metagenomics followed by assembly and gene calling on the assembled contigs. The assembly is typically conducted either by individually assembling each sample or by co-assembling reads from all the samples. The co-assembly approach can potentially recover genes that display too low abundance to be assembled from individual samples. On the other hand, combining samples increases the risk of mixing data from closely related strains, which can hamper the assembly process. In this respect, assembly on individual samples followed by clustering of (near) identical genes is preferable. Thus, both approaches have potential pros and cons, but it remains to be evaluated which assembly strategy is most effective. Here, we have evaluated three assembly strategies for generating gene catalogues from metagenomes using a dataset of 124 samples from the Baltic Sea: (1) assembly on individual samples followed by clustering of the resulting genes, (2) co-assembly on all samples, and (3) mix assembly, combining individual and co-assembly.
Results: The mix-assembly approach resulted in a more extensive nonredundant gene set than the other approaches and with more genes predicted to be complete and that could be functionally annotated. The mix assembly consists of 67 million genes (Baltic Sea gene set, BAGS) that have been functionally and taxonomically annotated. The majority of the BAGS genes are dissimilar (< 95% amino acid identity) to the Tara Oceans gene dataset, and hence, BAGS represents a valuable resource for brackish water research.
Conclusion: The mix-assembly approach represents a feasible approach to increase the information obtained from metagenomic samples. Video abstract.
Keywords: Assembly approach; Baltic Sea; Brackish water; Gene catalogue; Metagenomics; Mix assembly.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures




References
-
- Sunagawa S, Coelho LP, Chaffron S, Kultima JR, Labadie K, Salazar G, et al. Structure and function of the global ocean microbiome. Science. 2015;348. American Association for the Advancement of Science. [cited 2021 Aug 11]. Available from: 10.1126/science.1261359. - PubMed
-
- Steinegger M. Ultrafast and sensitive sequence search and clustering methods in the era of next generation sequencing [Internet]. Technische Universität München; 2018. Available from: http://mediatum.ub.tum.de/doc/1435187/678546.pdf.
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Miscellaneous
