

Catching a killer: Mechanisms of programmed cell death and immune activation in Amyotrophic Lateral Sclerosis
- PMID: 35524757
- PMCID: PMC9489610
- DOI: 10.1111/imr.13083
Catching a killer: Mechanisms of programmed cell death and immune activation in Amyotrophic Lateral Sclerosis
Abstract
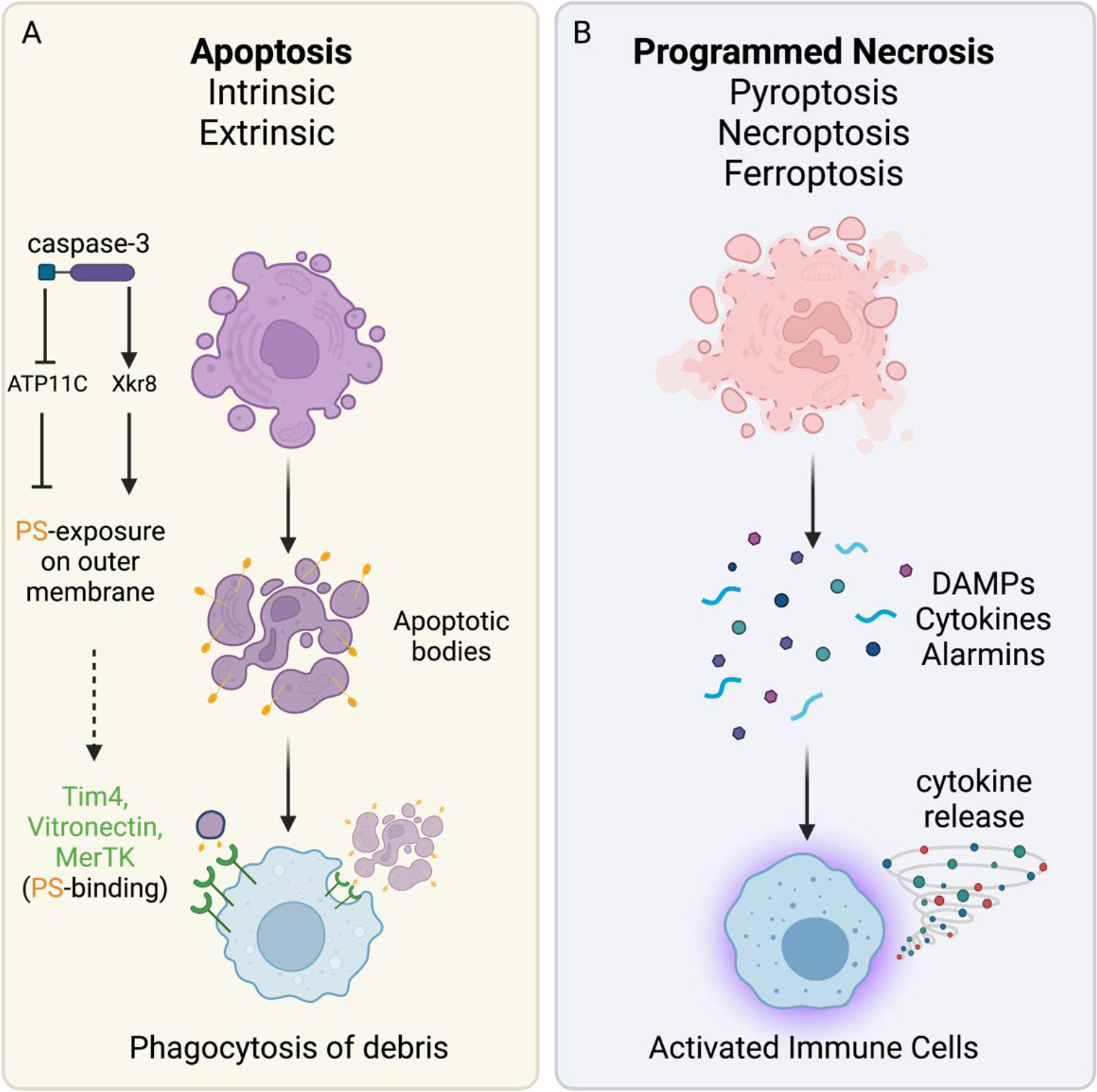
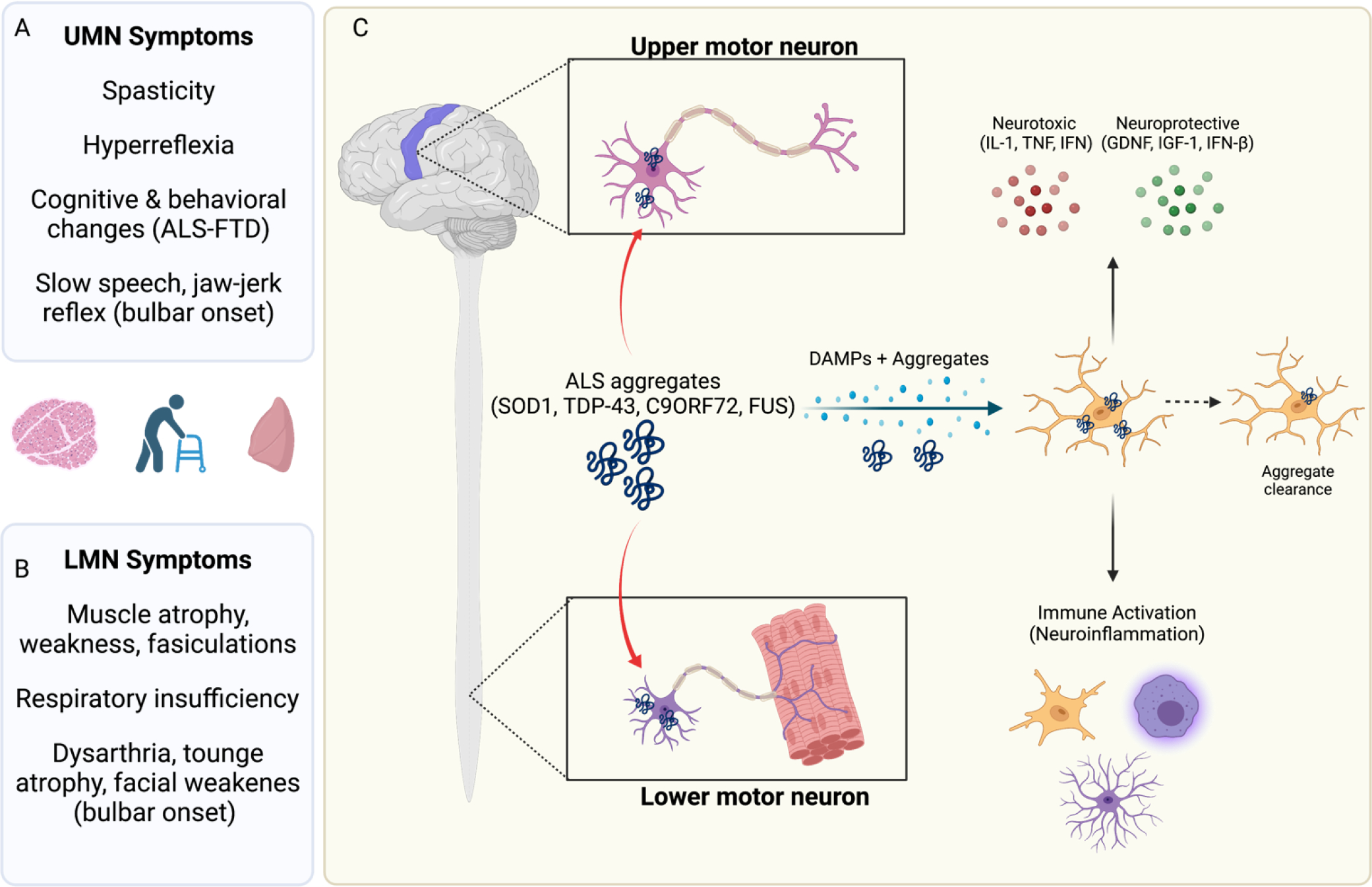
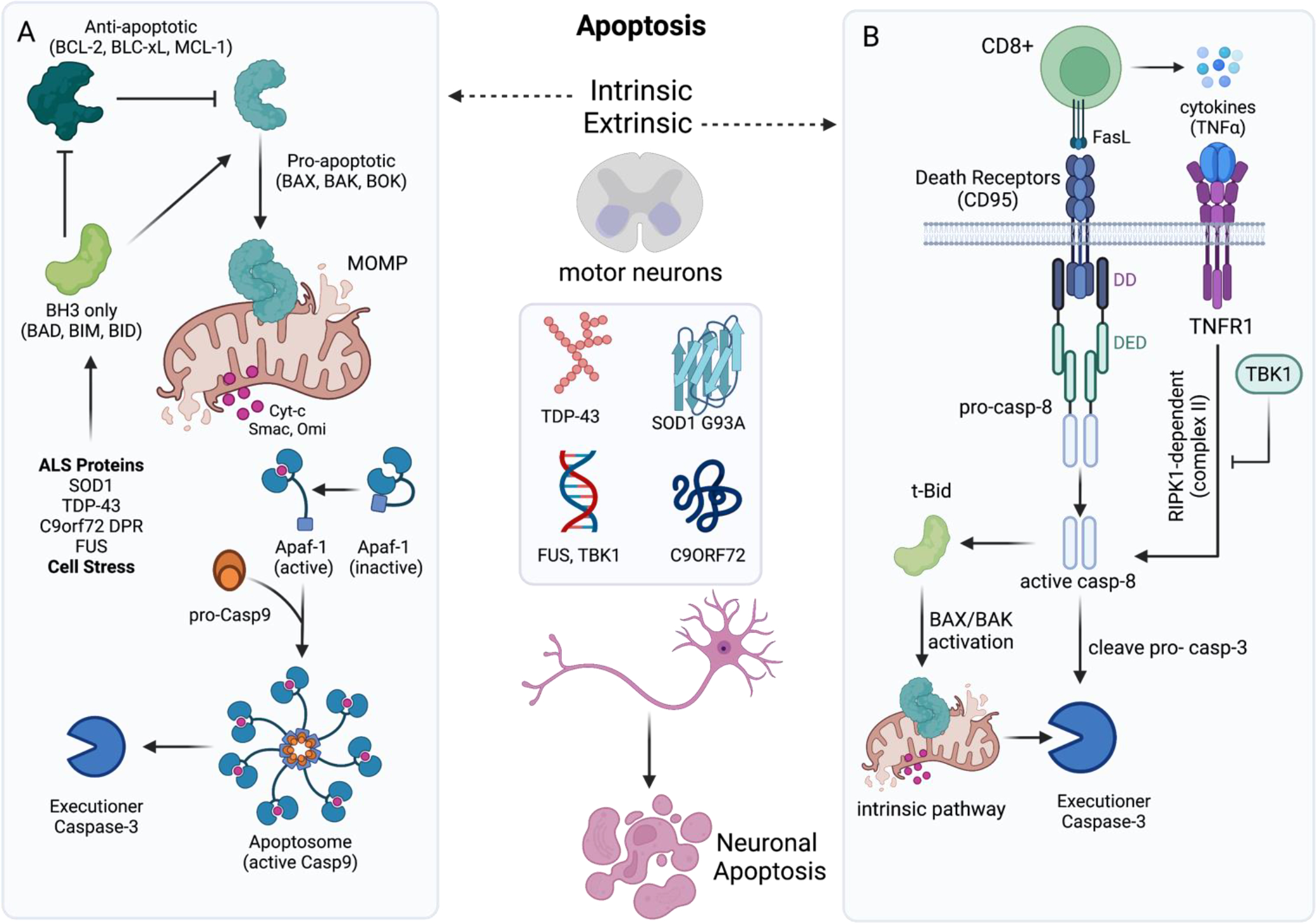
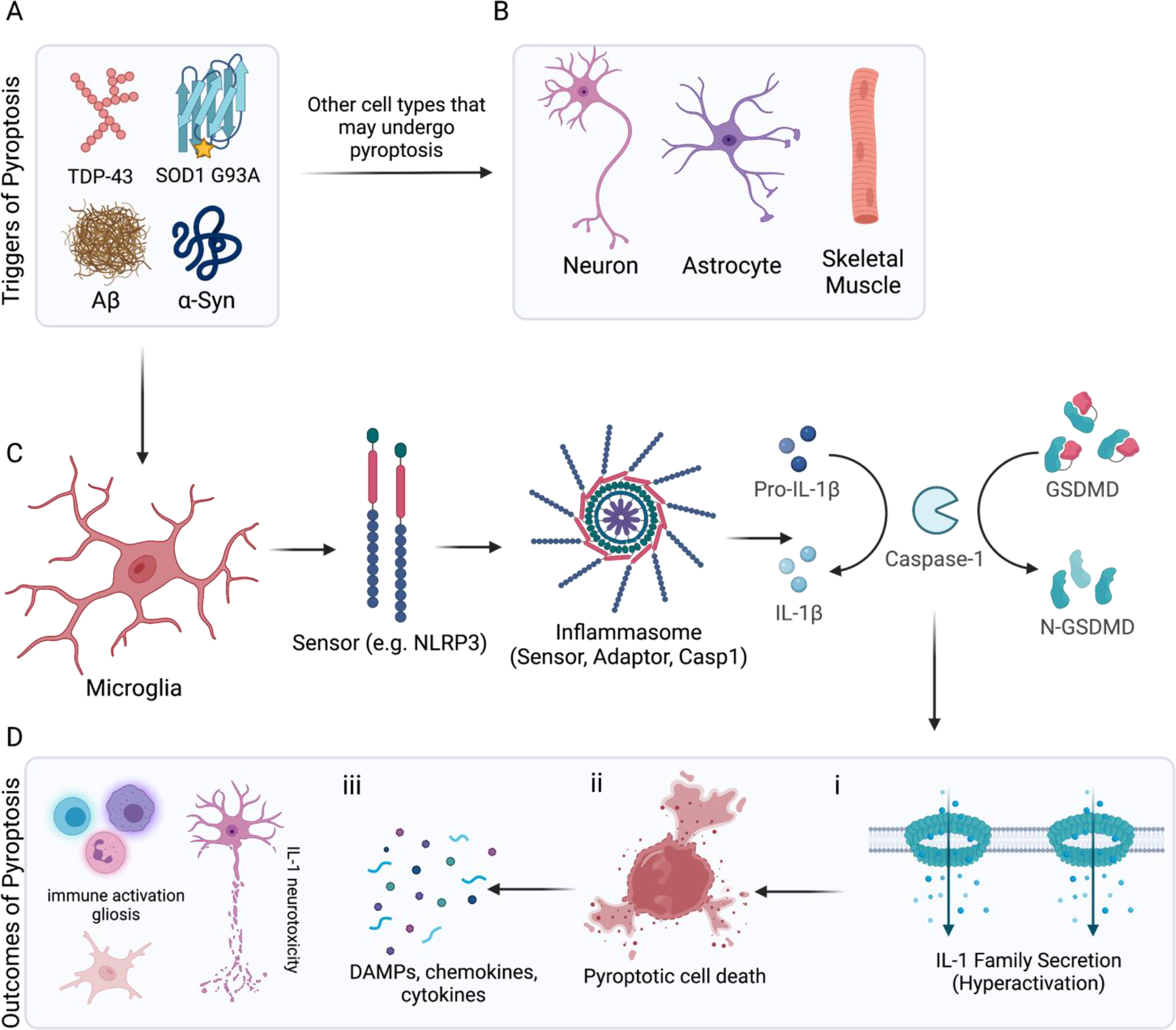
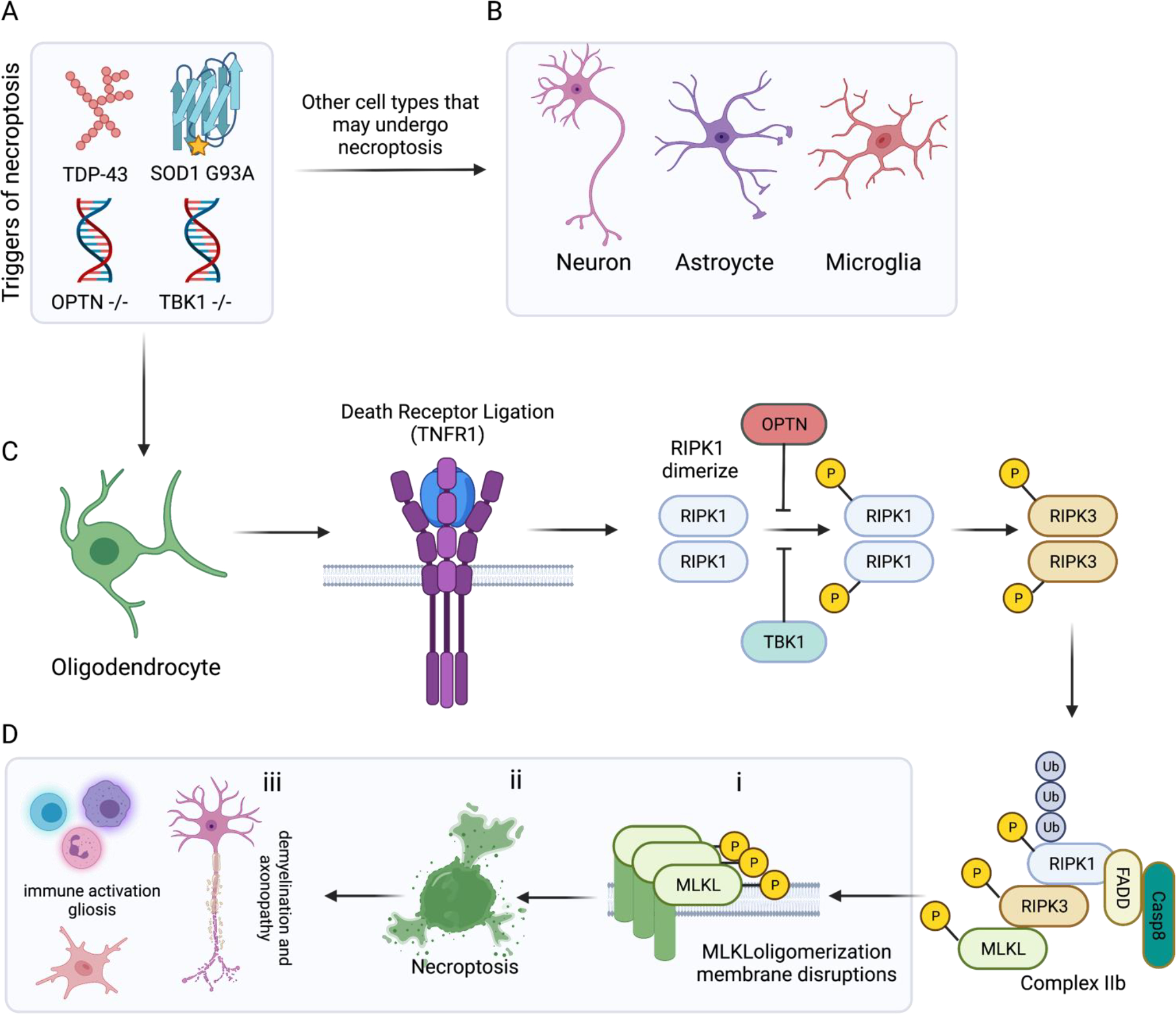
In the central nervous system (CNS), execution of programmed cell death (PCD) is crucial for proper neurodevelopment. However, aberrant activation of these pathways in adult CNS leads to neurodegenerative diseases including amyotrophic lateral sclerosis (ALS). How a cell dies is critical, as it can drive local immune activation and tissue damage. Classical apoptosis engages several mechanisms to evoke "immunologically silent" responses, whereas other forms of programmed death such as pyroptosis, necroptosis, and ferroptosis release molecules that can potentiate immune responses and inflammation. In ALS, a fatal neuromuscular disorder marked by progressive death of lower and upper motor neurons, several cell types in the CNS express machinery for multiple PCD pathways. The specific cell types engaging PCD, and ultimate mechanisms by which neuronal death occurs in ALS are not well defined. Here, we provide an overview of different PCD pathways implicated in ALS. We also examine immune activation in ALS and differentiate apoptosis from necrotic mechanisms based on downstream immunological consequences. Lastly, we highlight therapeutic strategies that target cell death pathways in the treatment of neurodegeneration and inflammation in ALS.
Keywords: cell death; innate immunology; neurodegeneration.
© 2022 John Wiley & Sons A/S. Published by John Wiley & Sons Ltd.
Conflict of interest statement
Declaration of Interests:
I.M.C. serves on scientific advisory boards of GSK pharmaceuticals and Limm therapeutics. His lab receives research support from Abbvie/Allergan pharmaceuticals.
Figures
References
-
- Abe K, Aoki M, Tsuji S, Itoyama Y, Sobue G, Togo M, Hamada C, Tanaka M, Akimoto M, Nakamura K, et al. (2017). Safety and efficacy of edaravone in well defined patients with amyotrophic lateral sclerosis: a randomised, double-blind, placebo-controlled trial. The Lancet Neurology 16, 505–512. 10.1016/S1474-4422(17)30115-1. - DOI - PubMed
Publication types
MeSH terms
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Miscellaneous
