

BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia
- PMID: 35526014
- PMCID: PMC9077874
- DOI: 10.1186/s13024-022-00535-x
BIN1 is a key regulator of proinflammatory and neurodegeneration-related activation in microglia
Abstract
Background: The BIN1 locus contains the second-most significant genetic risk factor for late-onset Alzheimer's disease. BIN1 undergoes alternate splicing to generate tissue- and cell-type-specific BIN1 isoforms, which regulate membrane dynamics in a range of crucial cellular processes. Whilst the expression of BIN1 in the brain has been characterized in neurons and oligodendrocytes in detail, information regarding microglial BIN1 expression is mainly limited to large-scale transcriptomic and proteomic data. Notably, BIN1 protein expression and its functional roles in microglia, a cell type most relevant to Alzheimer's disease, have not been examined in depth.
Methods: Microglial BIN1 expression was analyzed by immunostaining mouse and human brain, as well as by immunoblot and RT-PCR assays of isolated microglia or human iPSC-derived microglial cells. Bin1 expression was ablated by siRNA knockdown in primary microglial cultures in vitro and Cre-lox mediated conditional deletion in adult mouse brain microglia in vivo. Regulation of neuroinflammatory microglial signatures by BIN1 in vitro and in vivo was characterized using NanoString gene panels and flow cytometry methods. The transcriptome data was explored by in silico pathway analysis and validated by complementary molecular approaches.
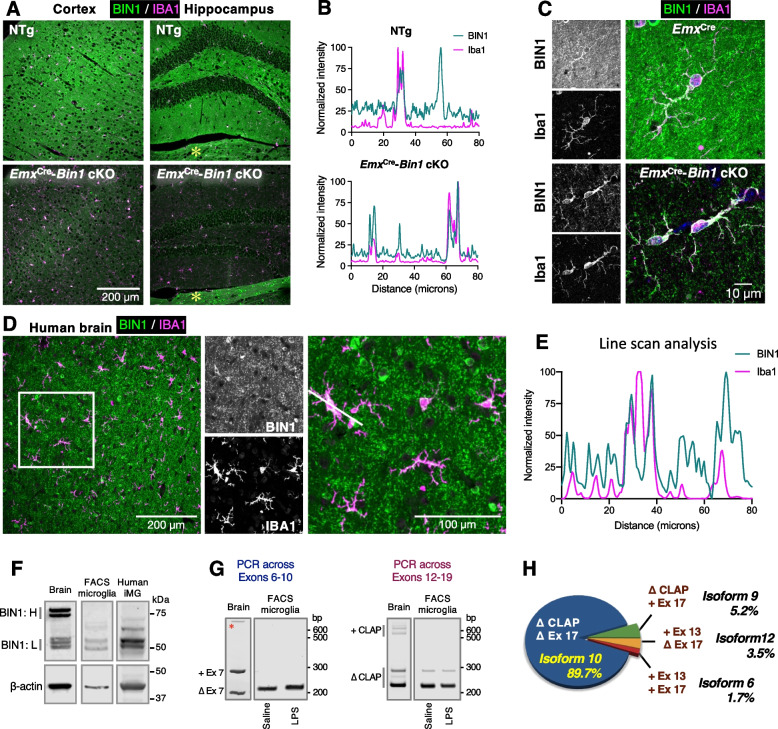
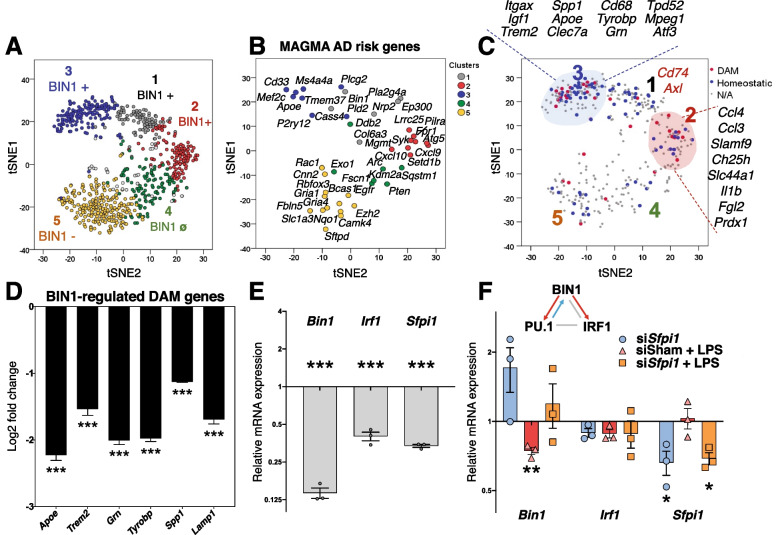
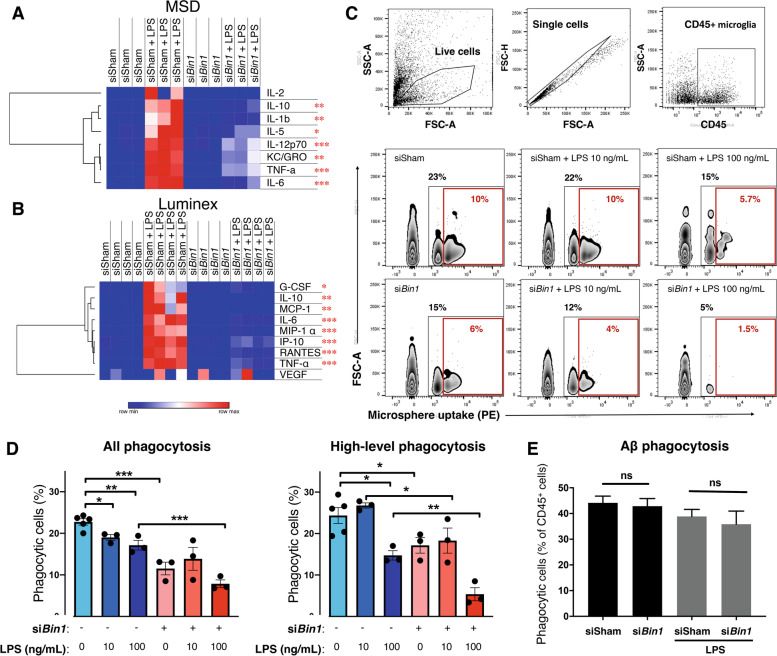
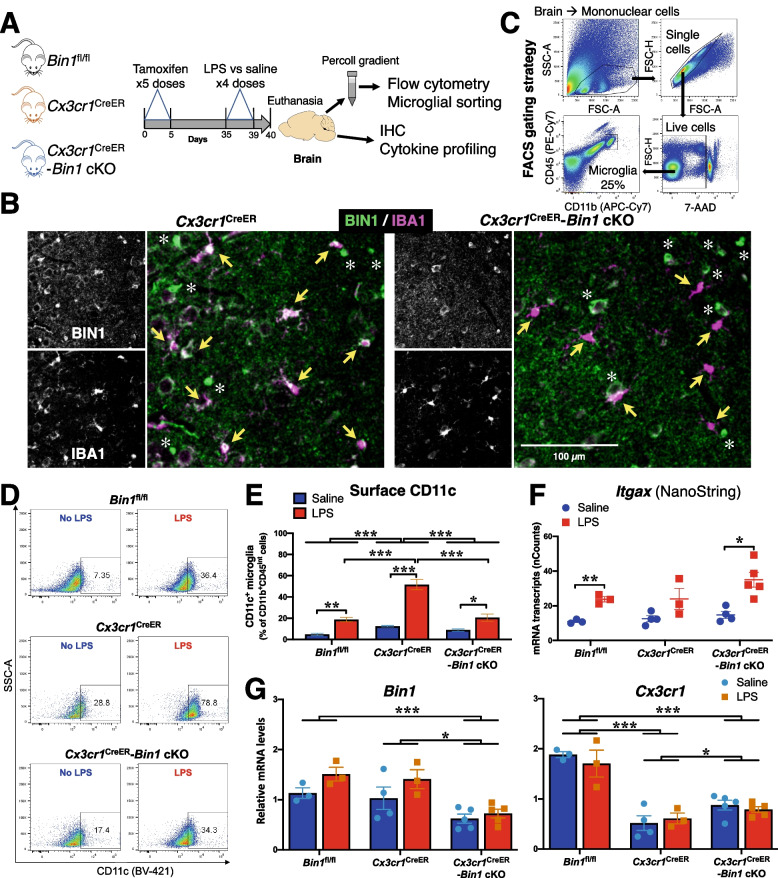
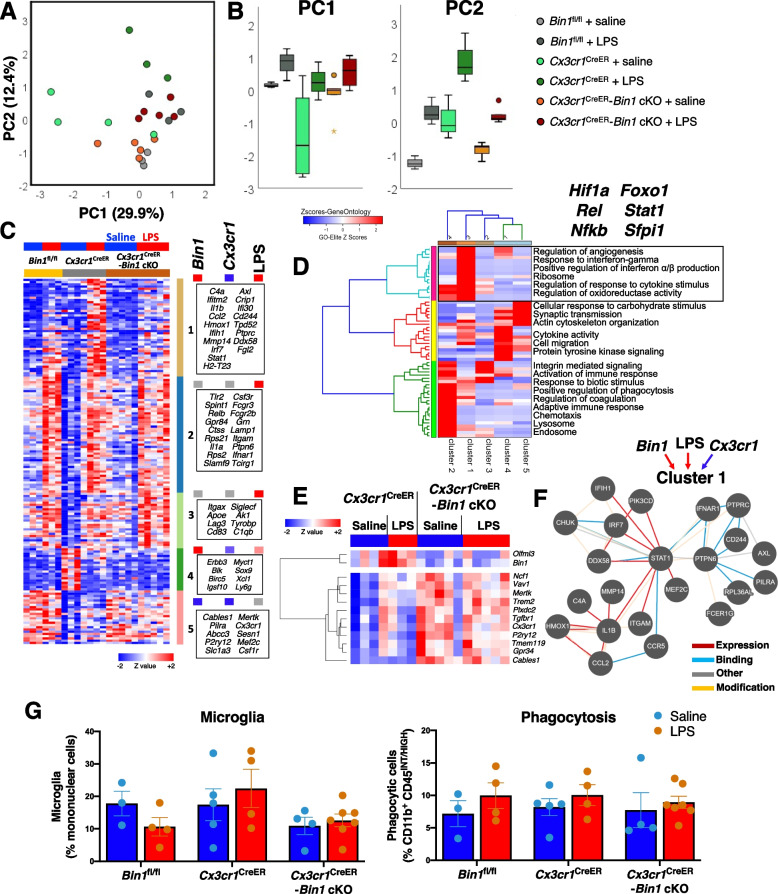
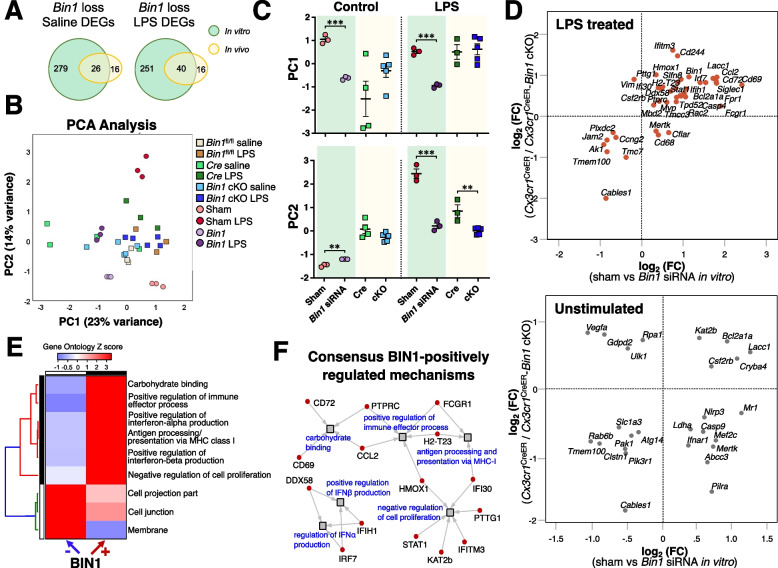
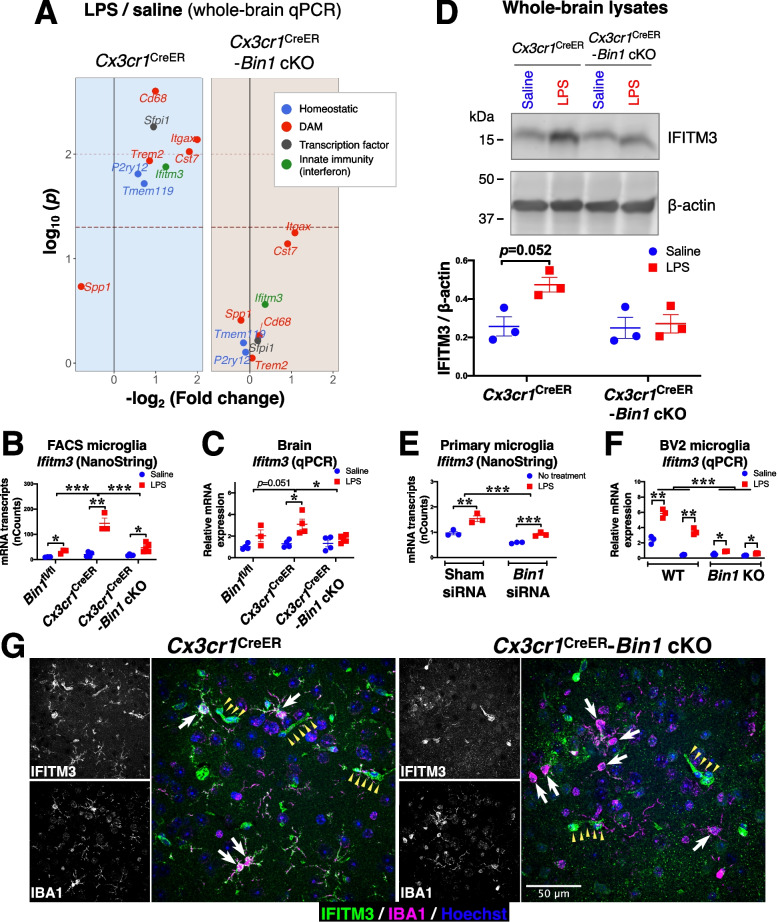
Results: Here, we characterized microglial BIN1 expression in vitro and in vivo and ascertained microglia expressed BIN1 isoforms. By silencing Bin1 expression in primary microglial cultures, we demonstrate that BIN1 regulates the activation of proinflammatory and disease-associated responses in microglia as measured by gene expression and cytokine production. Our transcriptomic profiling revealed key homeostatic and lipopolysaccharide (LPS)-induced inflammatory response pathways, as well as transcription factors PU.1 and IRF1 that are regulated by BIN1. Microglia-specific Bin1 conditional knockout in vivo revealed novel roles of BIN1 in regulating the expression of disease-associated genes while counteracting CX3CR1 signaling. The consensus from in vitro and in vivo findings showed that loss of Bin1 impaired the ability of microglia to mount type 1 interferon responses to proinflammatory challenge, particularly the upregulation of a critical type 1 immune response gene, Ifitm3.
Conclusions: Our convergent findings provide novel insights into microglial BIN1 function and demonstrate an essential role of microglial BIN1 in regulating brain inflammatory response and microglial phenotypic changes. Moreover, for the first time, our study shows a regulatory relationship between Bin1 and Ifitm3, two Alzheimer's disease-related genes in microglia. The requirement for BIN1 to regulate Ifitm3 upregulation during inflammation has important implications for inflammatory responses during the pathogenesis and progression of many neurodegenerative diseases.
Keywords: Alzheimer’s disease; BIN1; CX3CR1; GWAS risk factor; IFITM3; IRF1; IRF7; Innate immunity; LPS; Microglia; Neuroinflammation; PU.1.
© 2022. The Author(s).
Conflict of interest statement
The authors declare that they have no competing interests.
Figures

References
-
- Prokic I, Cowling BS, Laporte J. Amphiphysin 2 (BIN1) in physiology and diseases. J Mol Med. 2014;92:453–463. - PubMed
Publication types
MeSH terms
Substances
Grants and funding
LinkOut - more resources
Full Text Sources
Medical
Molecular Biology Databases
