

Endothelial Cell Phenotype, a Major Determinant of Venous Thrombo-Inflammation
- PMID: 35528838
- PMCID: PMC9068971
- DOI: 10.3389/fcvm.2022.864735
Endothelial Cell Phenotype, a Major Determinant of Venous Thrombo-Inflammation
Abstract
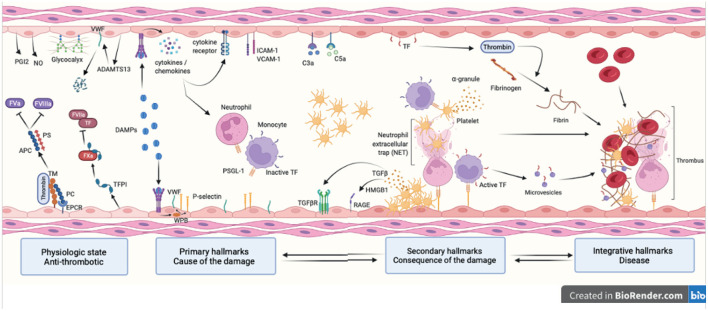
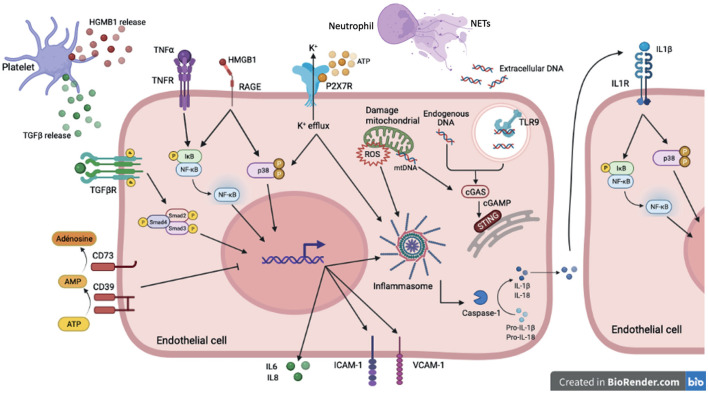
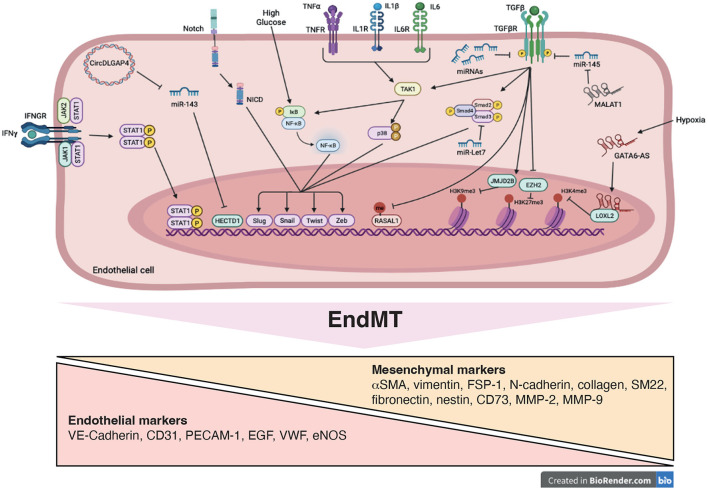
Reduced blood flow velocity in the vein triggers inflammation and is associated with the release into the extracellular space of alarmins or damage-associated molecular patterns (DAMPs). These molecules include extracellular nucleic acids, extracellular purinergic nucleotides (ATP, ADP), cytokines and extracellular HMGB1. They are recognized as a danger signal by immune cells, platelets and endothelial cells. Hence, endothelial cells are capable of sensing environmental cues through a wide variety of receptors expressed at the plasma membrane. The endothelium is then responding by expressing pro-coagulant proteins, including tissue factor, and inflammatory molecules such as cytokines and chemokines involved in the recruitment and activation of platelets and leukocytes. This ultimately leads to thrombosis, which is an active pro-inflammatory process, tightly regulated, that needs to be properly resolved to avoid further vascular damages. These mechanisms are often dysregulated, which promote fibrinolysis defects, activation of the immune system and irreversible vascular damages further contributing to thrombotic and inflammatory processes. The concept of thrombo-inflammation is now widely used to describe the complex interactions between the coagulation and inflammation in various cardiovascular diseases. In endothelial cells, activating signals converge to multiple intracellular pathways leading to phenotypical changes turning them into inflammatory-like cells. Accumulating evidence suggest that endothelial to mesenchymal transition (EndMT) may be a major mechanism of endothelial dysfunction induced during inflammation and thrombosis. EndMT is a biological process where endothelial cells lose their endothelial characteristics and acquire mesenchymal markers and functions. Endothelial dysfunction might play a central role in orchestrating and amplifying thrombo-inflammation thought induction of EndMT processes. Mechanisms regulating endothelial dysfunction have been only partially uncovered in the context of thrombotic diseases. In the present review, we focus on the importance of the endothelial phenotype and discuss how endothelial plasticity may regulate the interplay between thrombosis and inflammation. We discuss how the endothelial cells are sensing and responding to environmental cues and contribute to thrombo-inflammation with a particular focus on venous thromboembolism (VTE). A better understanding of the precise mechanisms involved and the specific role of endothelial cells is needed to characterize VTE incidence and address the risk of recurrent VTE and its sequelae.
Keywords: endothelial cell; endothelial plasticity; fibrosis; inflammation; venous thromboembolism.
Copyright © 2022 Pilard, Ollivier, Gourdou-Latyszenok, Couturaud and Lemarié.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
Publication types
LinkOut - more resources
Full Text Sources