

Asymmetric Predictive Relationships Across Histone Modifications
- PMID: 35529103
- PMCID: PMC9075108
- DOI: 10.1038/s42256-022-00455-x
Asymmetric Predictive Relationships Across Histone Modifications
Abstract
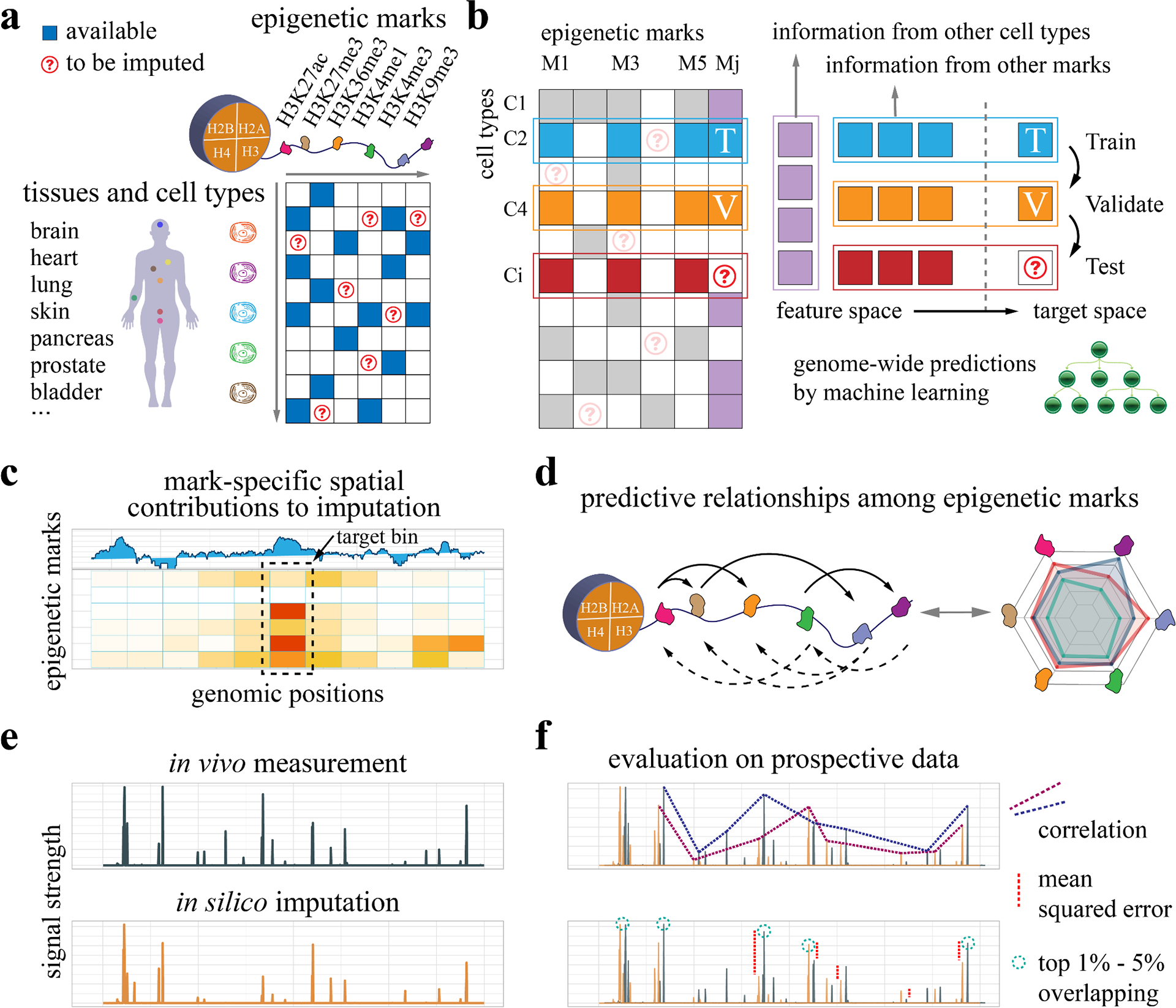
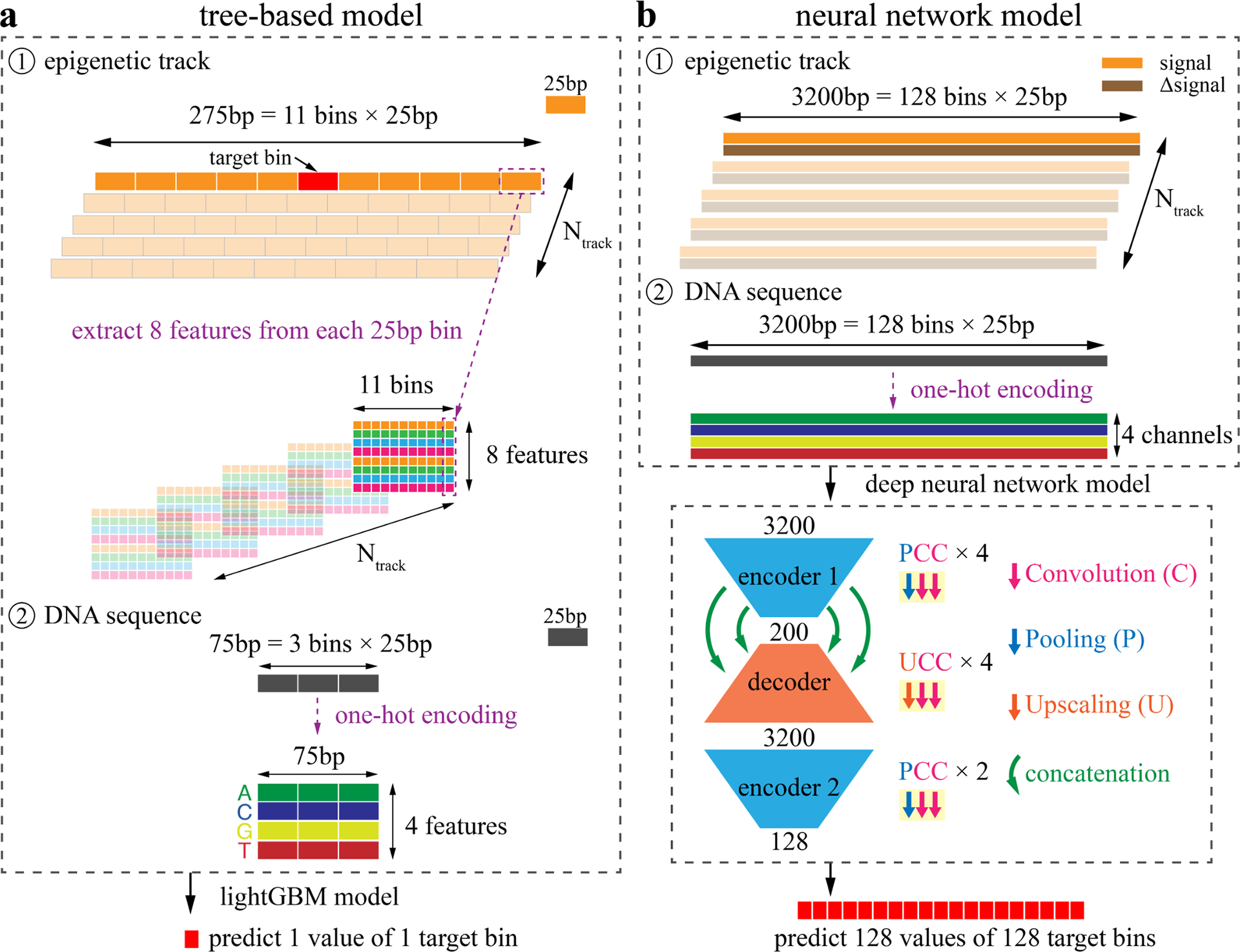
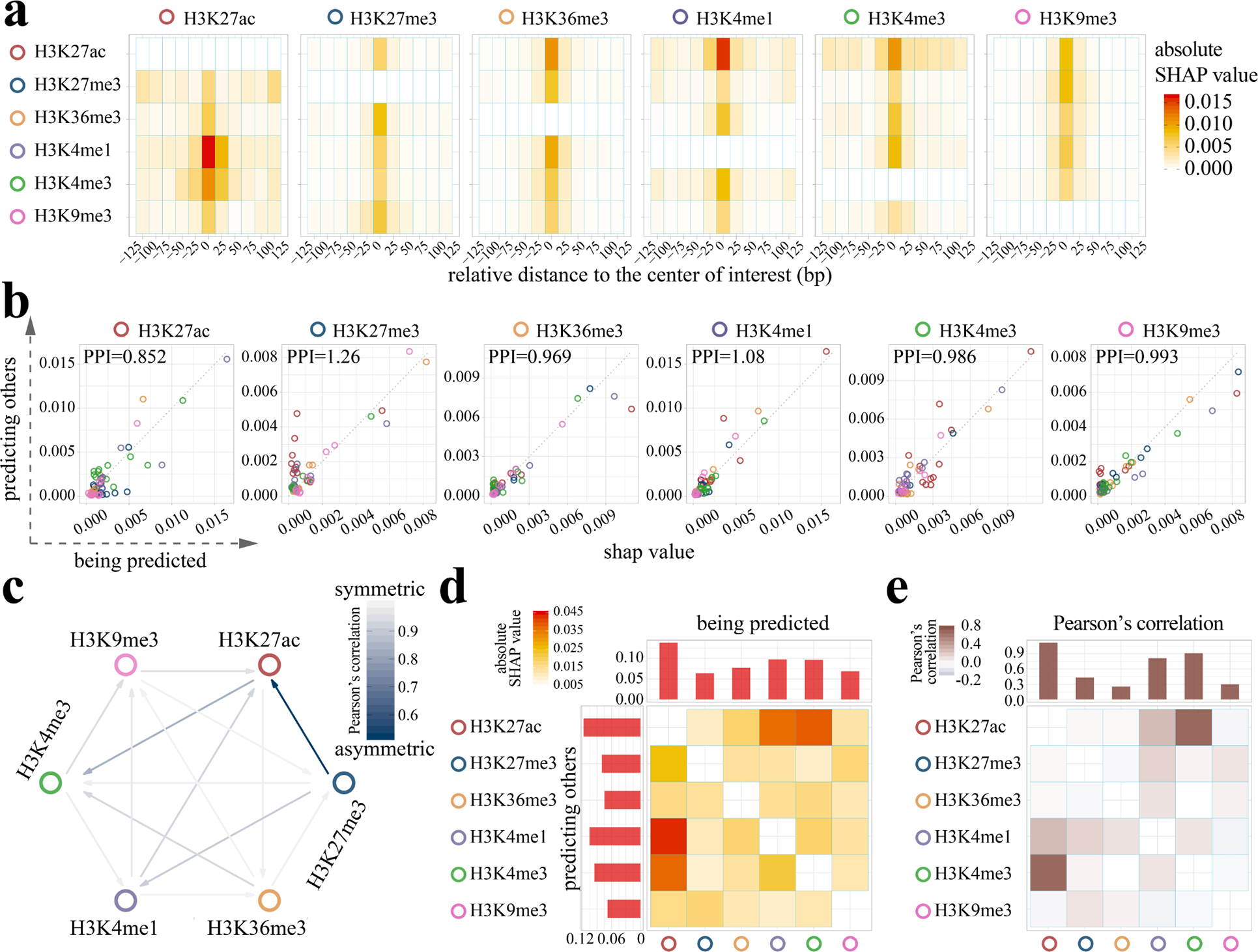
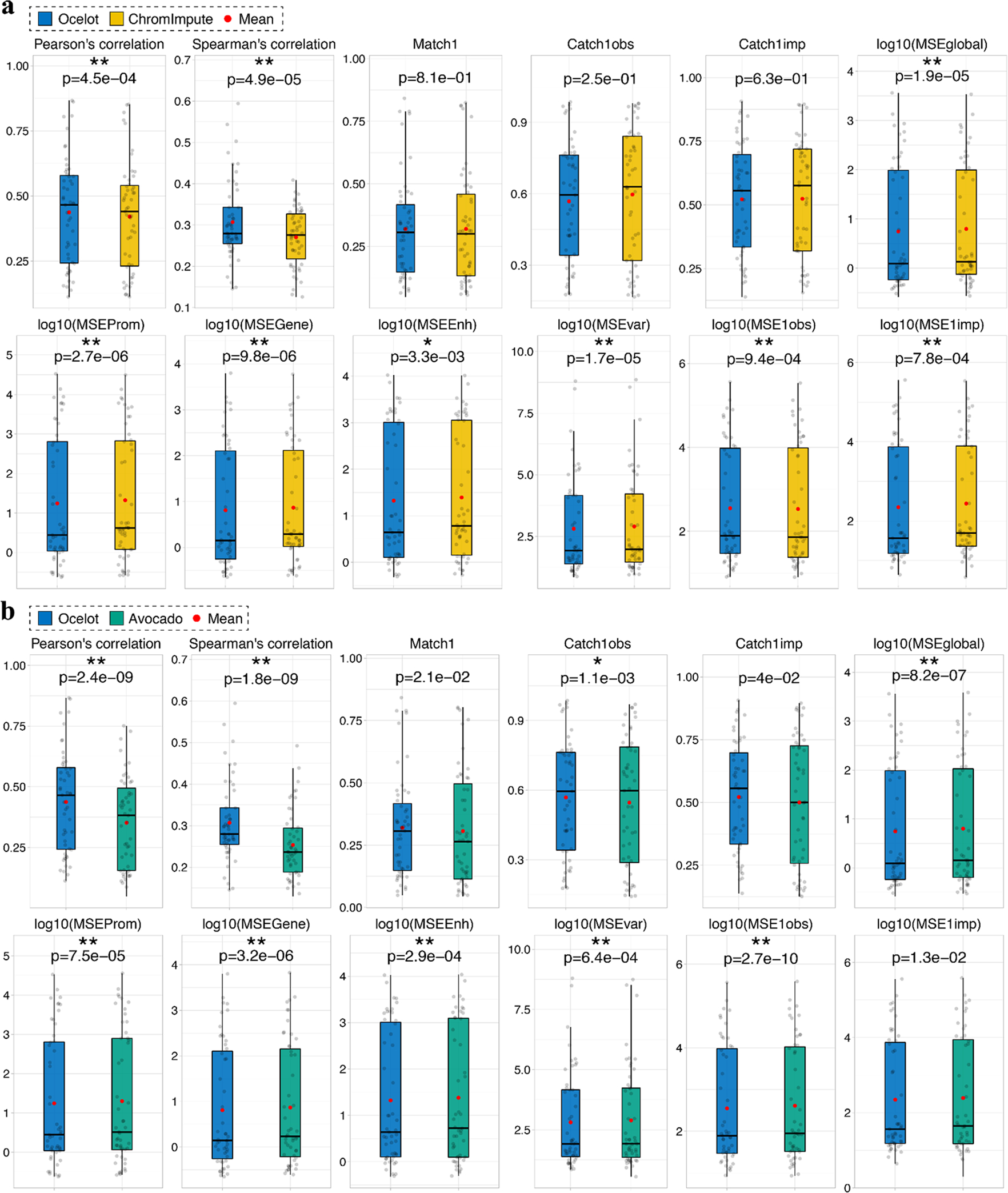
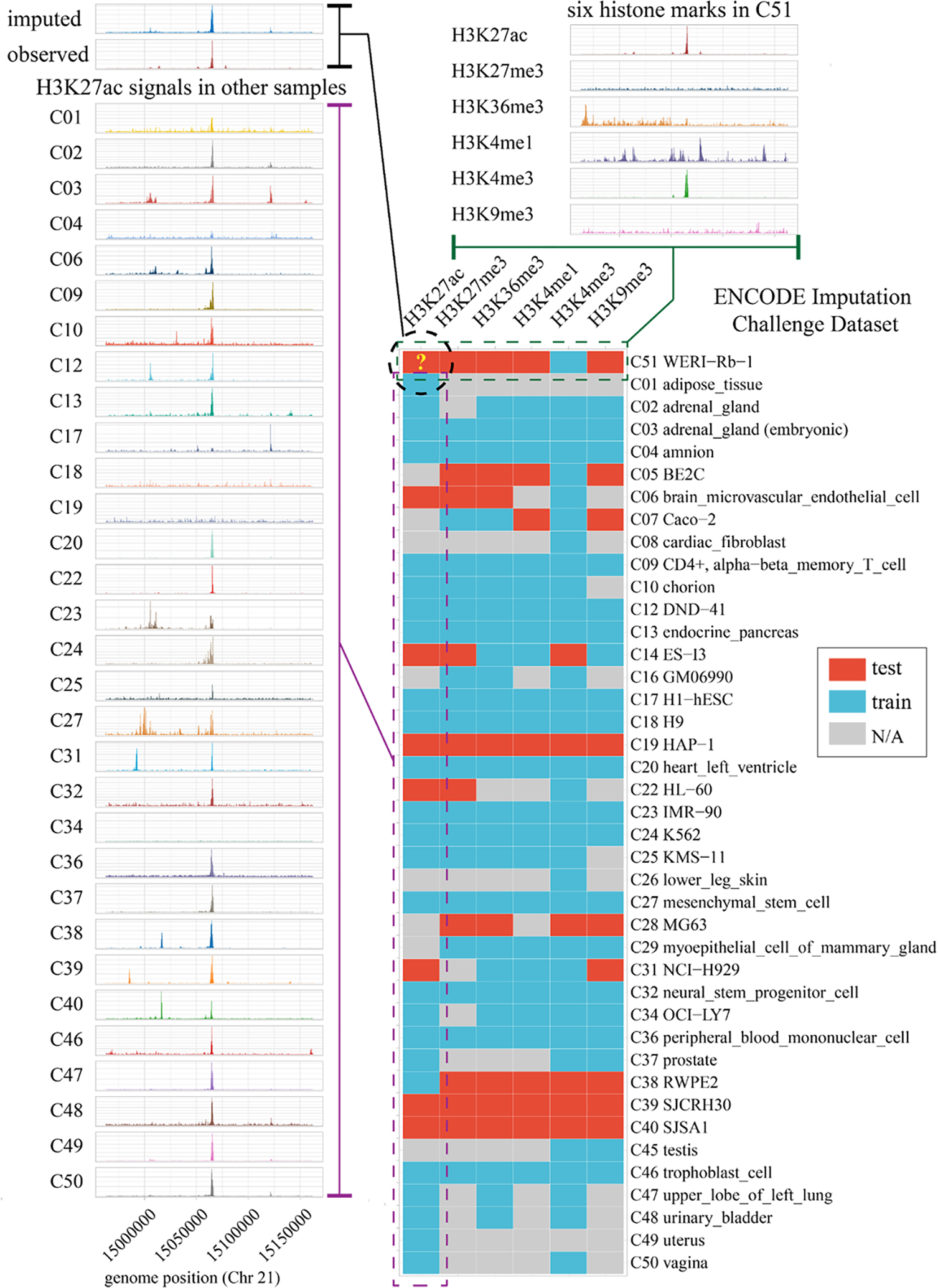
Decoding the epigenomic landscapes in diverse tissues and cell types is fundamental to understanding molecular mechanisms underlying many essential cellular processes and human diseases. Recent advances in artificial intelligence provide new methods and strategies for imputing unknown epigenomes based on existing data, yet how to reveal the predictive relationships among epigenetic marks remains largely unexplored. Here we present a machine learning approach for epigenomic imputation and interpretation. Through dissection of the spatial contributions from six histone marks, we reveal the prevalent and asymmetric cross-prediction relationships among these marks. Meanwhile, our approach achieved high predictive performance on held-out prospective epigenomes and outperformed the state-of-the-art. To facilitate future research, we further applied this approach to impute a total of 527 and 2,455 unavailable genome-wide histone modification signal tracks for the ENCODE3 and Roadmap datasets, respectively.
Keywords: Epigenome; Histone Modification; Machine Learning.
Figures

Similar articles
-
A deep learning approach to automate whole-genome prediction of diverse epigenomic modifications in plants.New Phytol. 2021 Oct;232(2):880-897. doi: 10.1111/nph.17630. Epub 2021 Aug 12. New Phytol. 2021. PMID: 34287908
-
Large-scale imputation of epigenomic datasets for systematic annotation of diverse human tissues.Nat Biotechnol. 2015 Apr;33(4):364-76. doi: 10.1038/nbt.3157. Epub 2015 Feb 18. Nat Biotechnol. 2015. PMID: 25690853 Free PMC article.
-
Deciphering epigenomic code for cell differentiation using deep learning.BMC Genomics. 2019 Sep 12;20(1):709. doi: 10.1186/s12864-019-6072-8. BMC Genomics. 2019. PMID: 31510916 Free PMC article.
-
Histone Modification Landscapes as a Roadmap for Malaria Parasite Development.Front Cell Dev Biol. 2022 Apr 1;10:848797. doi: 10.3389/fcell.2022.848797. eCollection 2022. Front Cell Dev Biol. 2022. PMID: 35433676 Free PMC article. Review.
-
Pharmaco-epigenomics: On the Road of Translation Medicine.Adv Exp Med Biol. 2019;1168:31-42. doi: 10.1007/978-3-030-24100-1_3. Adv Exp Med Biol. 2019. PMID: 31713163 Review.
Cited by
-
Multi-omics based and AI-driven drug repositioning for epigenetic therapy in female malignancies.J Transl Med. 2025 Jul 25;23(1):837. doi: 10.1186/s12967-025-06856-x. J Transl Med. 2025. PMID: 40713639 Free PMC article. Review.
-
Computational Methods Summarizing Mutational Patterns in Cancer: Promise and Limitations for Clinical Applications.Cancers (Basel). 2023 Mar 24;15(7):1958. doi: 10.3390/cancers15071958. Cancers (Basel). 2023. PMID: 37046619 Free PMC article. Review.
-
JMnorm: a novel joint multi-feature normalization method for integrative and comparative epigenomics.Nucleic Acids Res. 2024 Jan 25;52(2):e11. doi: 10.1093/nar/gkad1146. Nucleic Acids Res. 2024. PMID: 38055833 Free PMC article.
References
-
- Venter JC et al. The sequence of the human genome. Science 291, 1304–1351 (2001). - PubMed
-
- Smith ZD & Meissner A DNA methylation: roles in mammalian development. Nat. Rev. Genet. 14, 204–220 (2013). - PubMed
-
- Wittkopp PJ & Kalay G Cis-regulatory elements: molecular mechanisms and evolutionary processes underlying divergence. Nat. Rev. Genet. 13, 59–69 (2011). - PubMed
Grants and funding
LinkOut - more resources
Full Text Sources