

Identification of Potent and Selective JAK1 Lead Compounds Through Ligand-Based Drug Design Approaches
- PMID: 35529449
- PMCID: PMC9068899
- DOI: 10.3389/fphar.2022.837369
Identification of Potent and Selective JAK1 Lead Compounds Through Ligand-Based Drug Design Approaches
Abstract
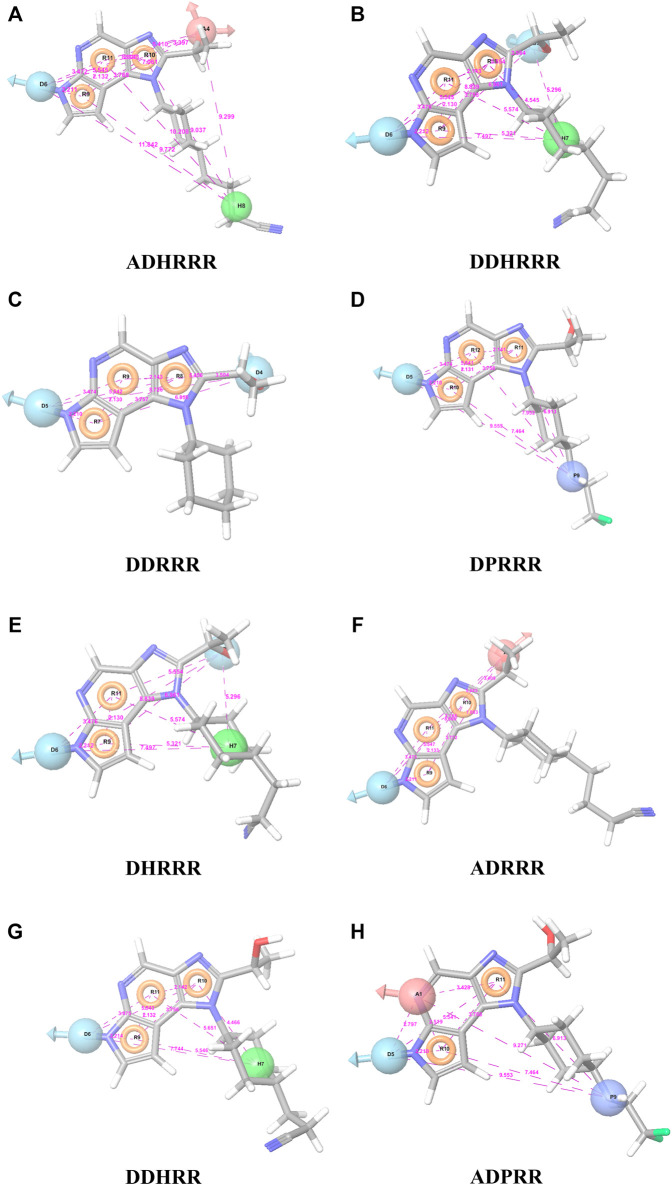
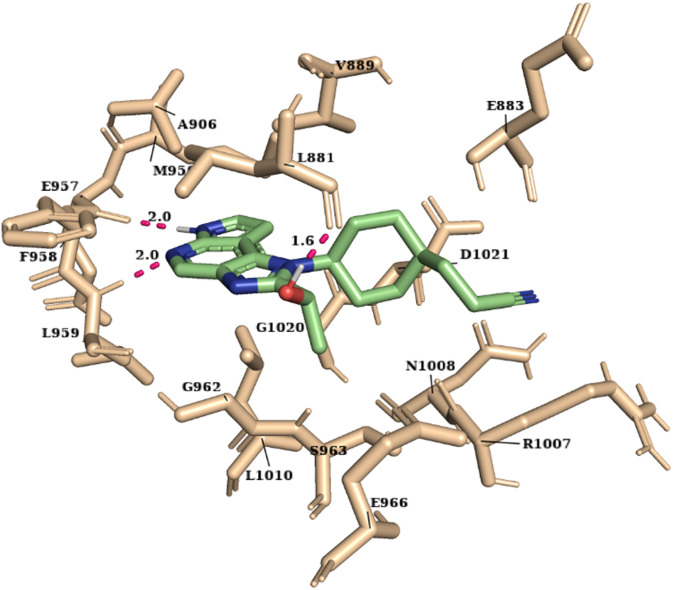
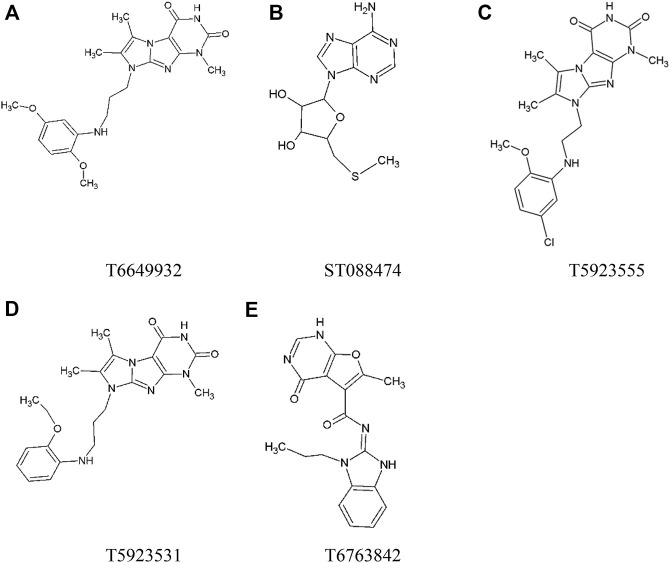
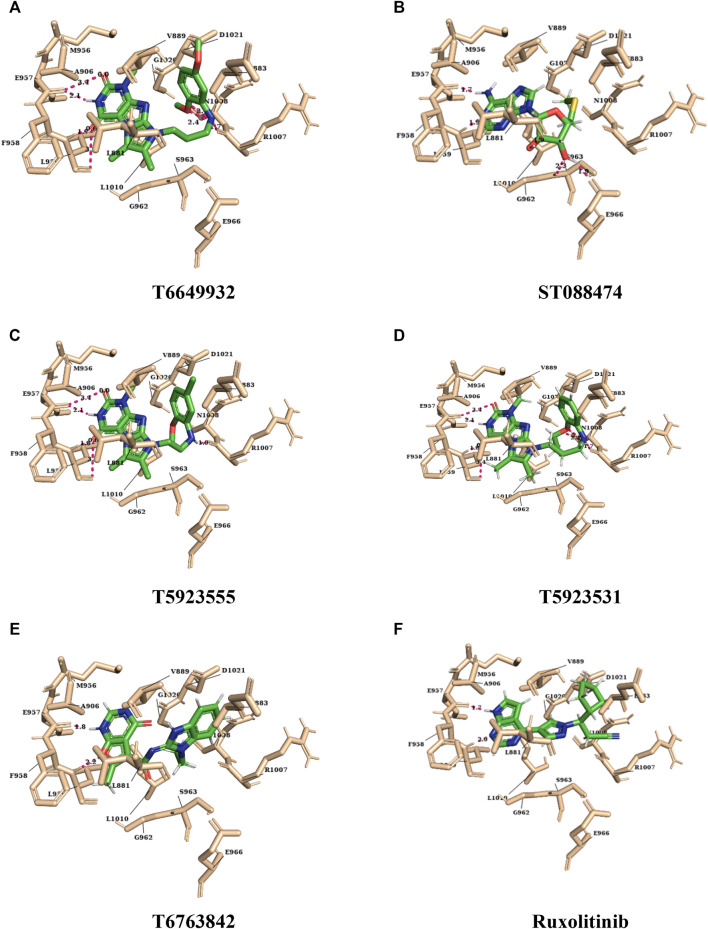
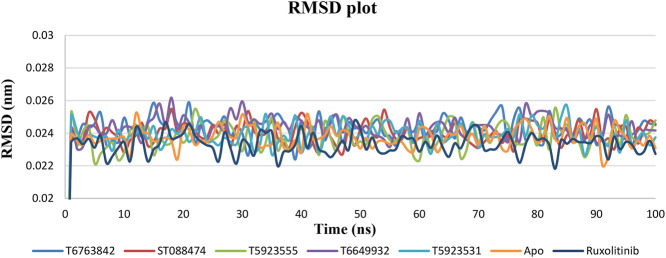
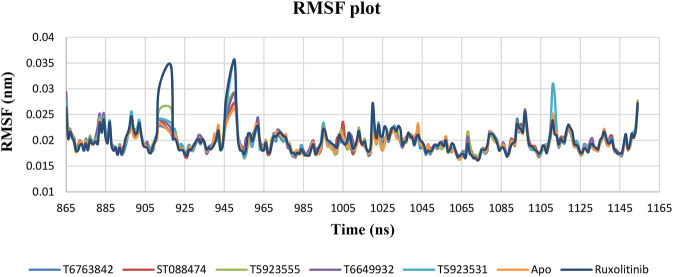
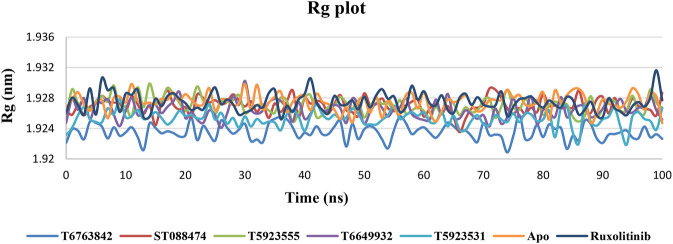
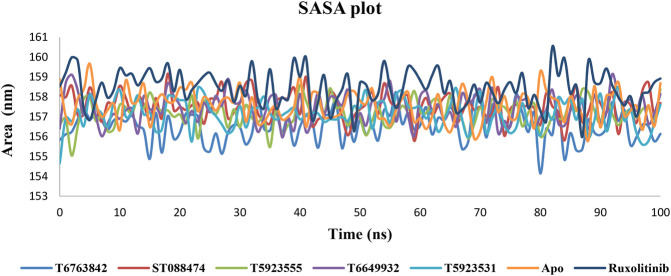
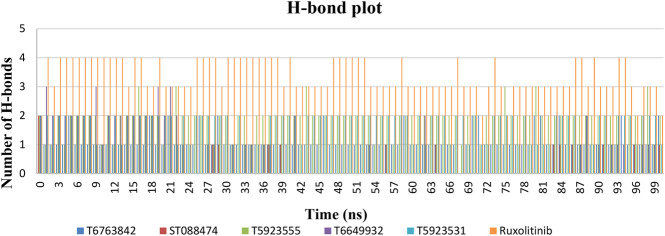
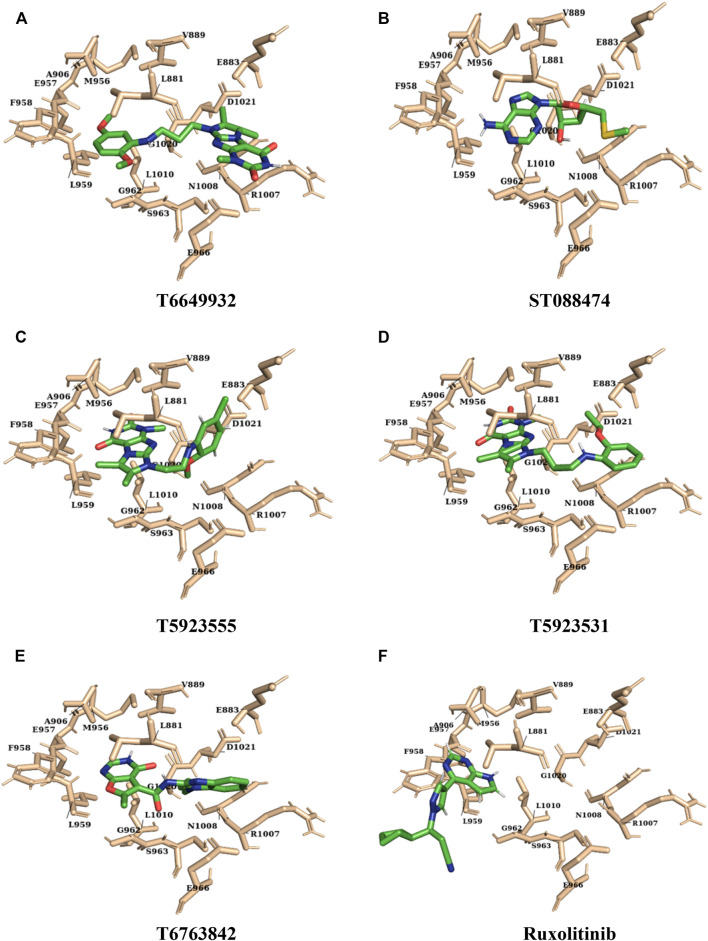
JAK1 plays a significant role in the intracellular signaling by interacting with cytokine receptors in different types of cells and is linked to the pathogenesis of various cancers and in the pathology of the immune system. In this study, ligand-based pharmacophore modeling combined with virtual screening and molecular docking methods was incorporated to identify the potent and selective lead compounds for JAK1. Initially, the ligand-based pharmacophore models were generated using a set of 52 JAK1 inhibitors named C-2 methyl/hydroxyethyl imidazopyrrolopyridines derivatives. Twenty-seven pharmacophore models with five and six pharmacophore features were generated and validated using potency and selectivity validation methods. During potency validation, the Guner-Henry score was calculated to check the accuracy of the generated models, whereas in selectivity validation, the pharmacophore models that are capable of identifying selective JAK1 inhibitors were evaluated. Based on the validation results, the best pharmacophore models ADHRRR, DDHRRR, DDRRR, DPRRR, DHRRR, ADRRR, DDHRR, and ADPRR were selected and taken for virtual screening against the Maybridge, Asinex, Chemdiv, Enamine, Lifechemicals, and Zinc database to identify the new molecules with novel scaffold that can bind to JAK1. A total of 4,265 hits were identified from screening and checked for acceptable drug-like properties. A total of 2,856 hits were selected after ADME predictions and taken for Glide molecular docking to assess the accurate binding modes of the lead candidates. Ninety molecules were shortlisted based on binding energy and H-bond interactions with the important residues of JAK1. The docking results were authenticated by calculating binding free energy for protein-ligand complexes using the MM-GBSA calculation and induced fit docking methods. Subsequently, the cross-docking approach was carried out to recognize the selective JAK1 lead compounds. Finally, top five lead compounds that were potent and selective against JAK1 were selected and validated using molecular dynamics simulation. Besides, the density functional theory study was also carried out for the selected leads. Through various computational studies, we observed good potency and selectivity of these lead compounds when compared with the drug ruxolitinib. Compounds such as T5923555 and T5923531 were found to be the best and can be further validated using in vitro and in vivo methods.
Keywords: JAK1; density function theory; molecular docking; molecular dynamics simulation; pharmacophore modeling; virtual screening.
Copyright © 2022 Babu, Nagarajan, Sathish, Negi, Sohn and Madhavan.
Conflict of interest statement
The authors declare that the research was conducted in the absence of any commercial or financial relationships that could be construed as a potential conflict of interest.
Figures
References
-
- Abraham M. J., Murtola T., Schulz R., Páll S., Smith J. C., Hess B., et al. (2015). GROMACS: High Performance Molecular Simulations through Multi-Level Parallelism from Laptops to Supercomputers. SoftwareX 1-2, 19–25. 10.1016/j.softx.2015.06.001 - DOI
-
- Babu S., Kulkarni S. A., Sohn H., Madhavan T. (2015). Identification of Leads through In Silico Approaches Utilizing Benzylthio-1h-Benzo[d]imidazol-1-Yl Acetic Acid Derivatives: A Potent CRTh2 Antagonist. J. Mol. Struct. 1102, 25–41. 10.1016/j.molstruc.2015.08.031 - DOI
-
- Becke A. D. (1998). A New Inhomogeneity Parameter in Density-Functional Theory. J. Chem. Phys. 109 (6), 2092–2098. 10.1063/1.476722 - DOI
-
- Caspers N. L., Han S., Rajamohan F., Hoth L. R., Geoghegan K. F., Subashi T. A., et al. (2016). Development of a High-Throughput crystal Structure-Determination Platform for JAK1 Using a Novel Metal-Chelator Soaking System. Acta Crystallogr. F Struct. Biol. Commun. 72 (11), 840–845. 10.1107/S2053230X16016356 - DOI - PMC - PubMed
LinkOut - more resources
Full Text Sources
Research Materials
Miscellaneous