

Ruscogenin alleviates LPS-triggered pulmonary endothelial barrier dysfunction through targeting NMMHC IIA to modulate TLR4 signaling
- PMID: 35530141
- PMCID: PMC9069402
- DOI: 10.1016/j.apsb.2021.09.017
Ruscogenin alleviates LPS-triggered pulmonary endothelial barrier dysfunction through targeting NMMHC IIA to modulate TLR4 signaling
Erratum in
-
Erratum: Author correction to 'Ruscogenin alleviates LPS-triggered pulmonary endothelial barrier dysfunction through targeting NMMHC IIA to modulate TLR4 signaling' [Acta Pharmaceutica Sinica B 12 (2022) 1198-1212].Acta Pharm Sin B. 2022 Jul;12(7):3198-3199. doi: 10.1016/j.apsb.2022.04.004. Epub 2022 Apr 8. Acta Pharm Sin B. 2022. PMID: 35865100 Free PMC article.
Abstract
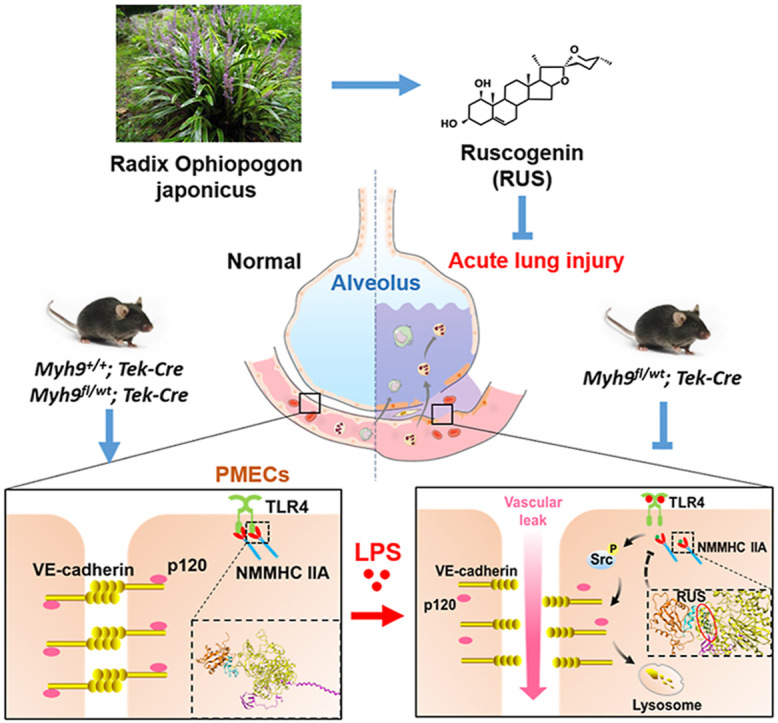
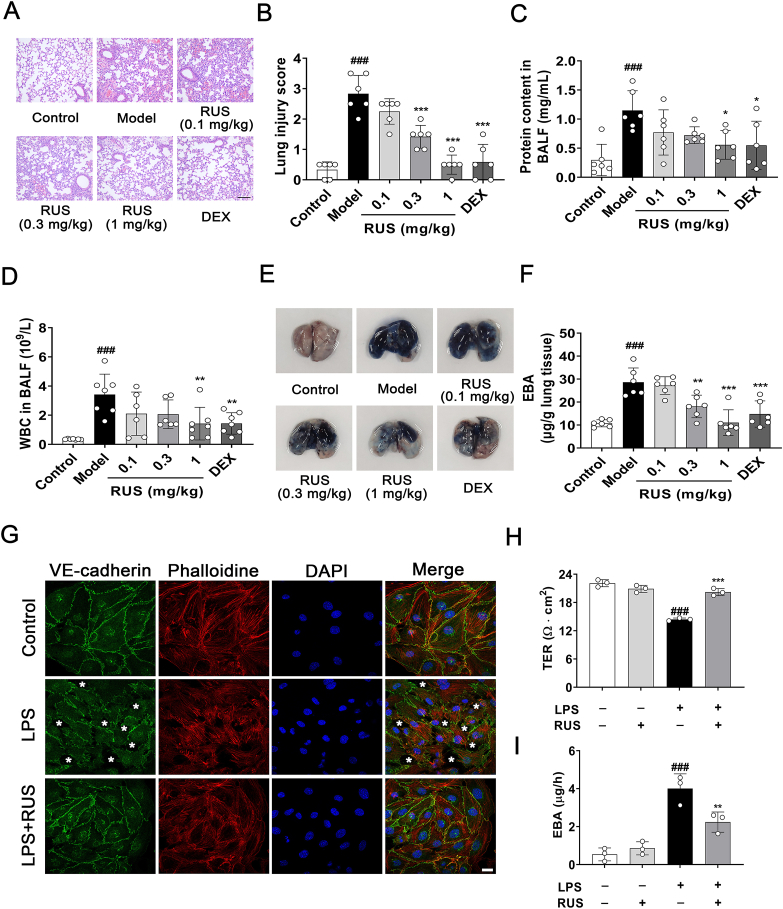
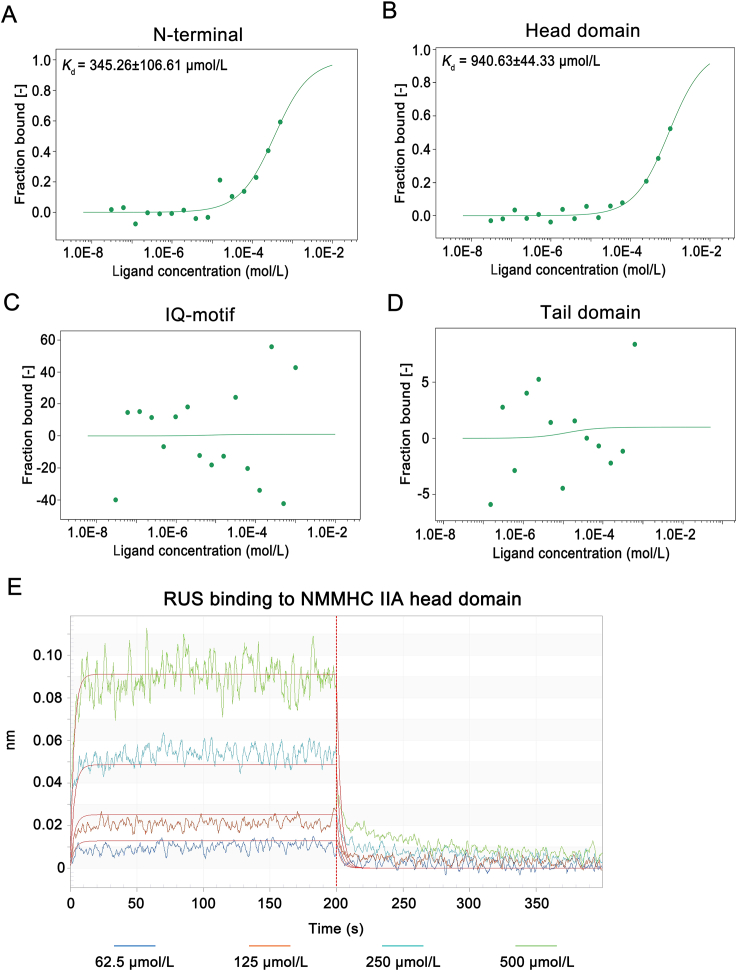
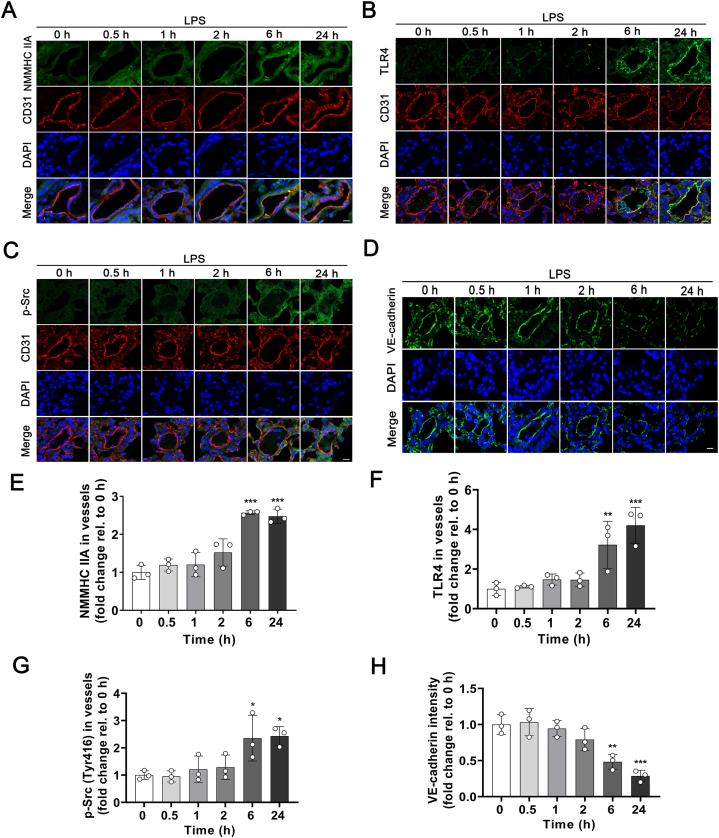
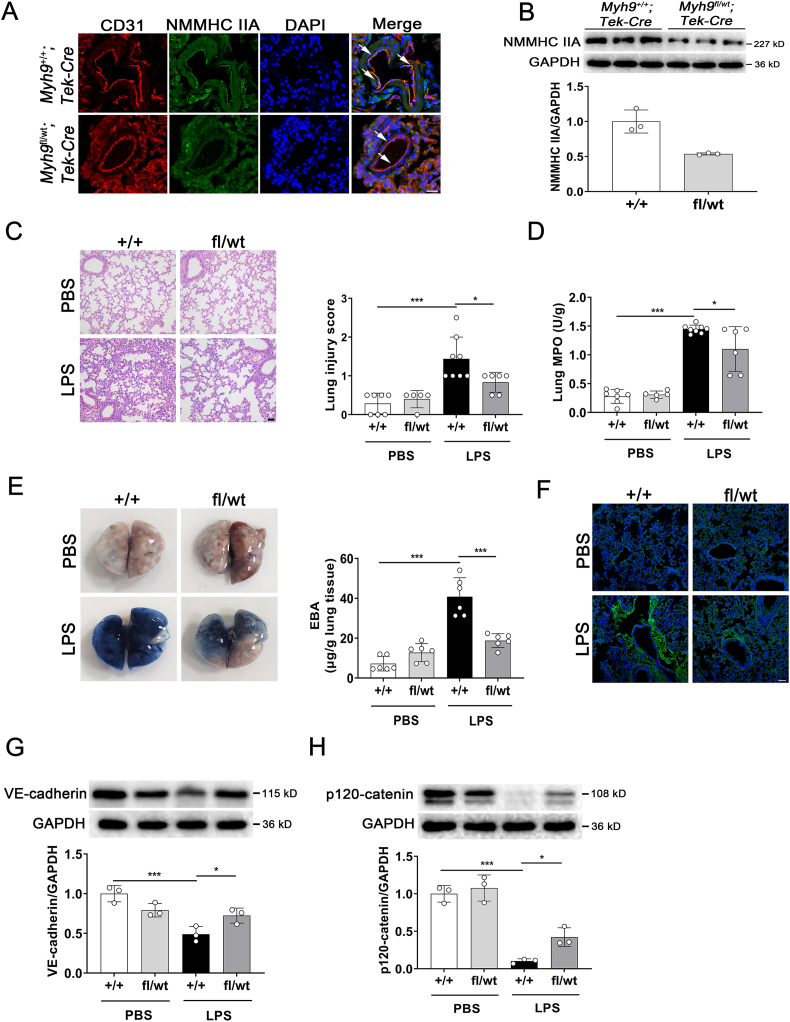
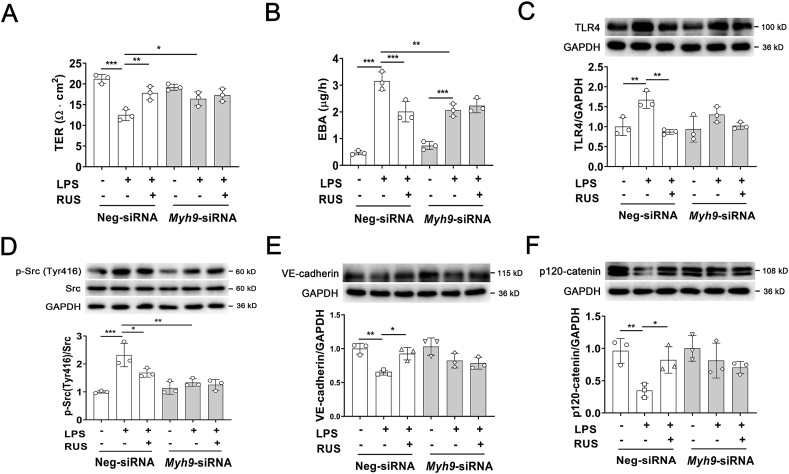
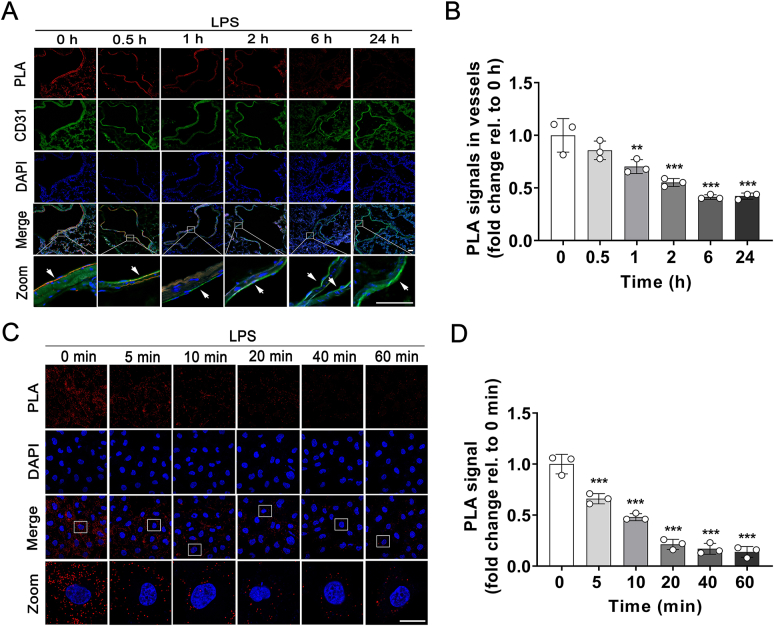
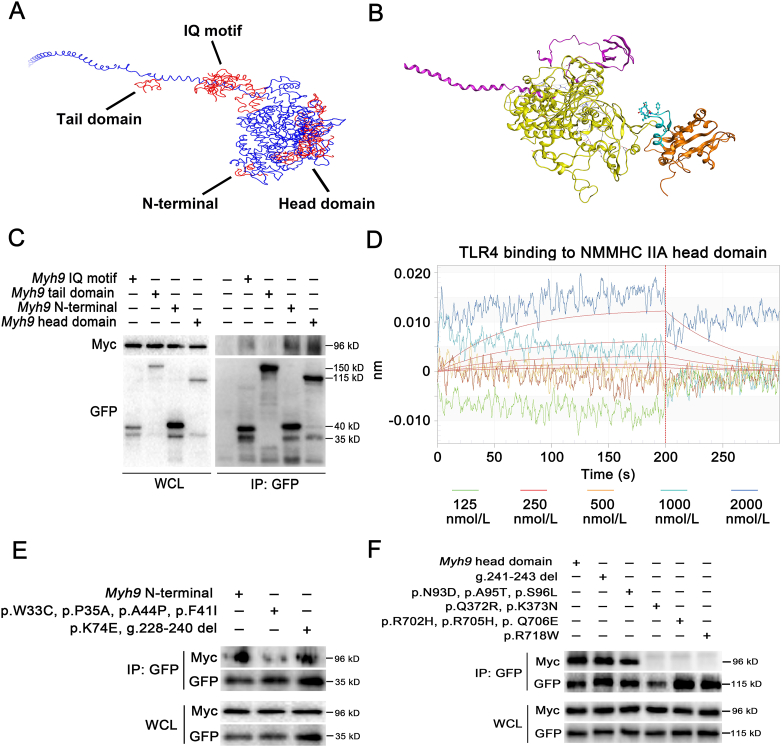
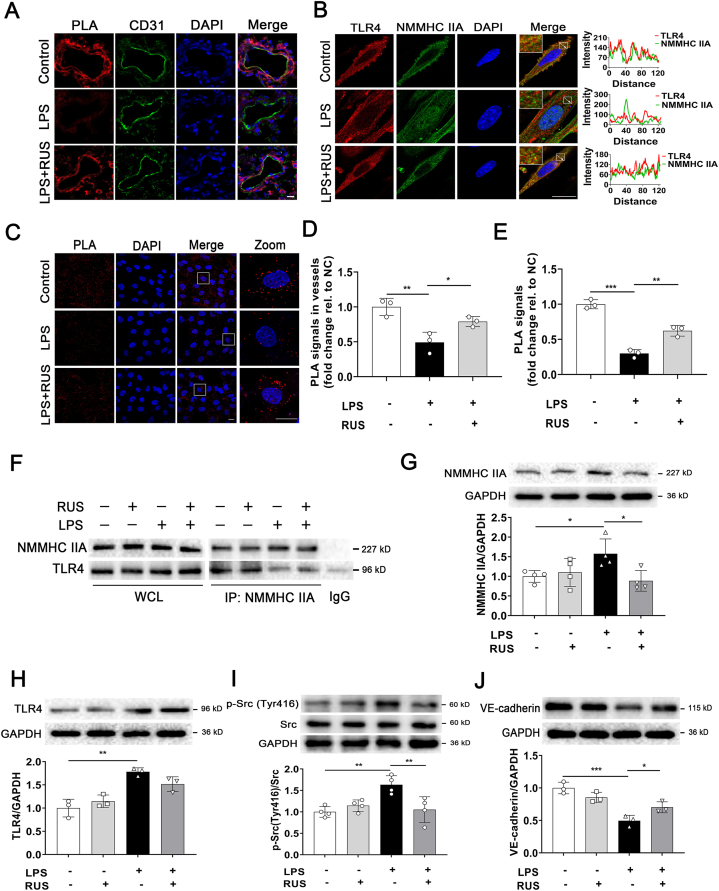
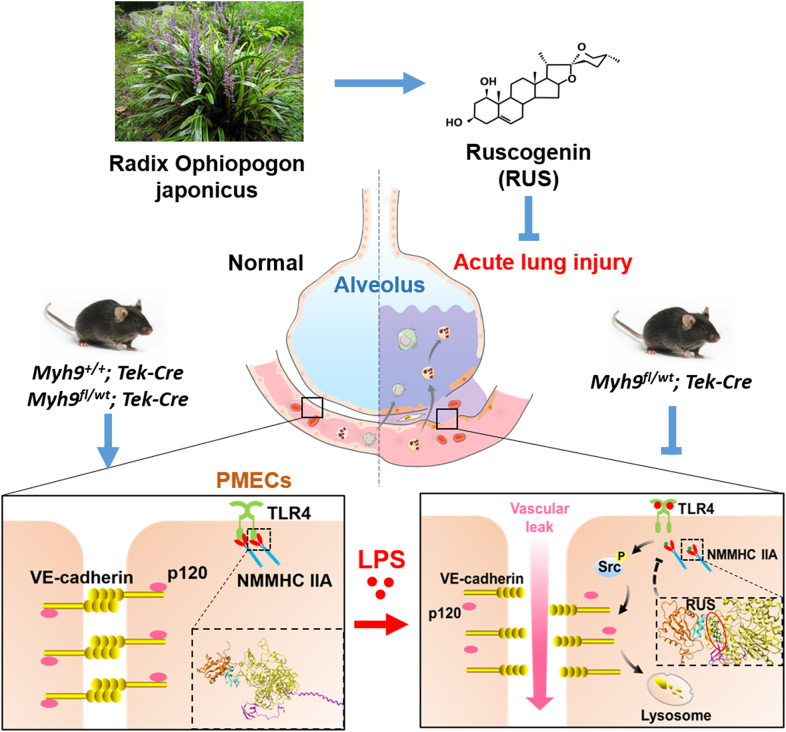
Pulmonary endothelial barrier dysfunction is a hallmark of clinical pulmonary edema and contributes to the development of acute lung injury (ALI). Here we reported that ruscogenin (RUS), an effective steroidal sapogenin of Radix Ophiopogon japonicus, attenuated lipopolysaccharides (LPS)-induced pulmonary endothelial barrier disruption through mediating non-muscle myosin heavy chain IIA (NMMHC IIA)‒Toll-like receptor 4 (TLR4) interactions. By in vivo and in vitro experiments, we observed that RUS administration significantly ameliorated LPS-triggered pulmonary endothelial barrier dysfunction and ALI. Moreover, we identified that RUS directly targeted NMMHC IIA on its N-terminal and head domain by serial affinity chromatography, molecular docking, biolayer interferometry, and microscale thermophoresis analyses. Downregulation of endothelial NMMHC IIA expression in vivo and in vitro abolished the protective effect of RUS. It was also observed that NMMHC IIA was dissociated from TLR4 and then activating TLR4 downstream Src/vascular endothelial cadherin (VE-cadherin) signaling in pulmonary vascular endothelial cells after LPS treatment, which could be restored by RUS. Collectively, these findings provide pharmacological evidence showing that RUS attenuates LPS-induced pulmonary endothelial barrier dysfunction by inhibiting TLR4/Src/VE-cadherin pathway through targeting NMMHC IIA and mediating NMMHC IIA‒TLR4 interactions.
Keywords: Acute lung injury; Endothelial barrier; Interaction; Lipopolysaccharide; Non-muscle myosin heavy chain IIA; Ruscogenin; TLR4; VE-cadherin.
© 2022 Chinese Pharmaceutical Association and Institute of Materia Medica, Chinese Academy of Medical Sciences. Production and hosting by Elsevier B.V.
Figures

References
-
- Thompson B.T., Chambers R.C., Liu K.D. Acute respiratory distress syndrome. N Engl J Med. 2017;377:562–572. - PubMed
-
- Ranieri V.M., Rubenfeld G.D., Thompson B.T., Ferguson N.D., Caldwell E., Fan E., et al. Acute respiratory distress syndrome: the Berlin Definition. J Am Med Assoc. 2012;307:2526–2533. - PubMed
-
- Millar F.R., Summers C., Griffiths M.J., Toshner M.R., Proudfoot A.G. The pulmonary endothelium in acute respiratory distress syndrome: insights and therapeutic opportunities. Thorax. 2016;71:462–473. - PubMed
LinkOut - more resources
Full Text Sources
Miscellaneous
