

Complex fitness landscape shapes variation in a hyperpolymorphic species
- PMID: 35532122
- PMCID: PMC9187340
- DOI: 10.7554/eLife.76073
Complex fitness landscape shapes variation in a hyperpolymorphic species
Abstract
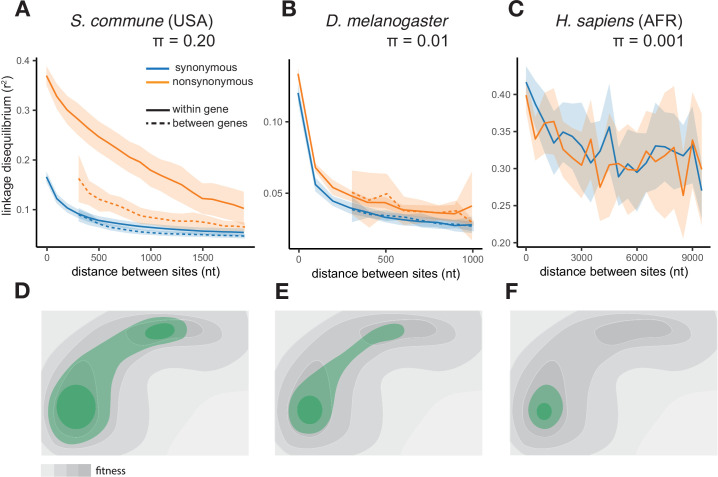
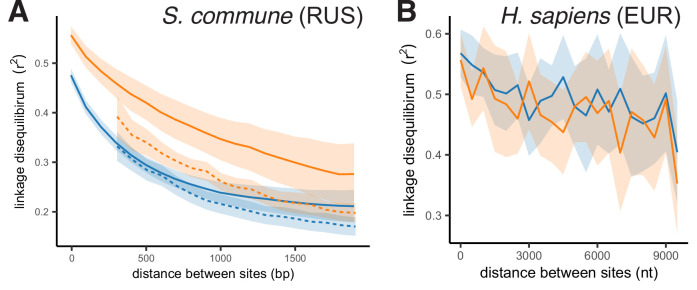
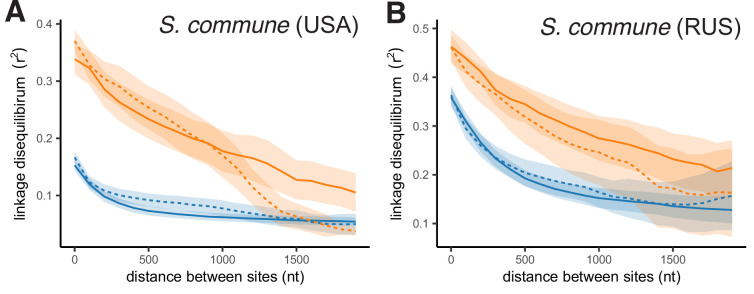
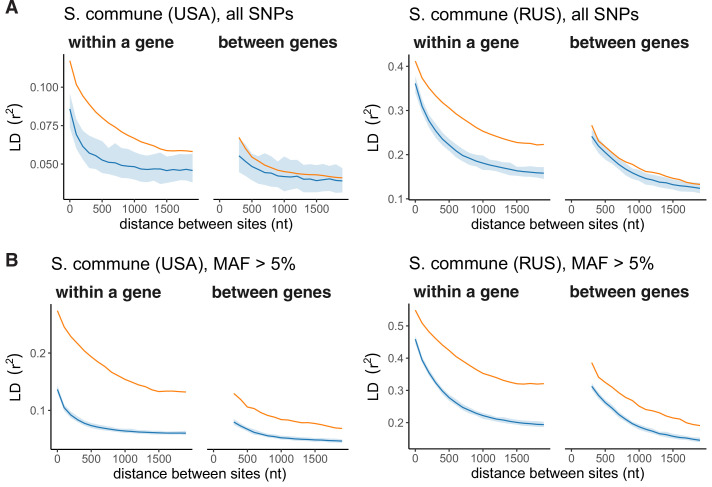
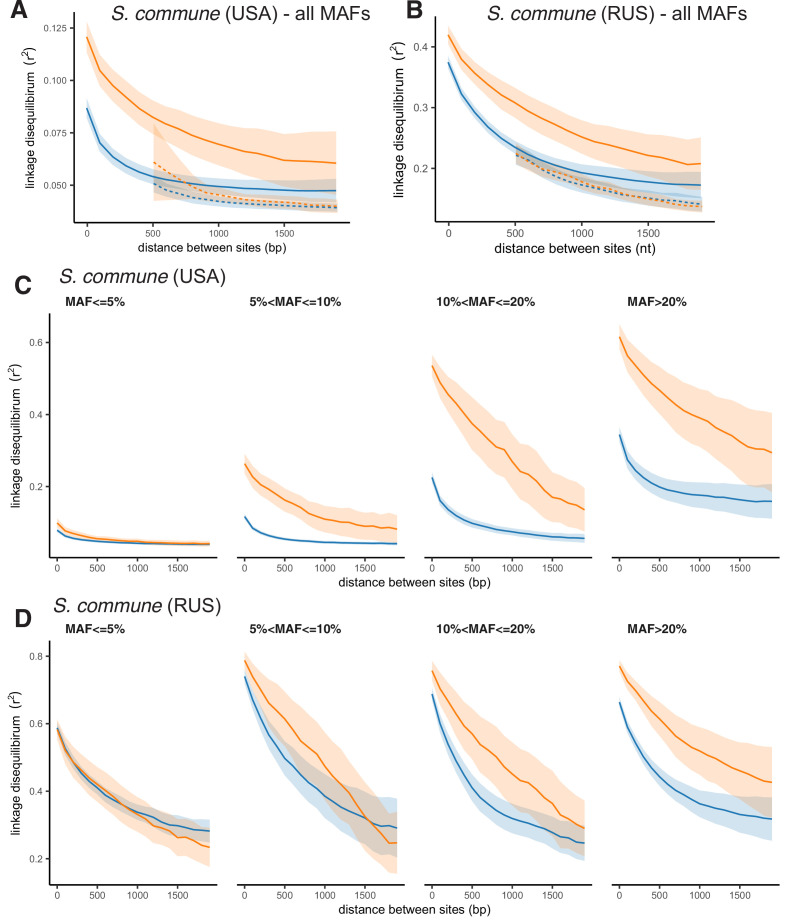
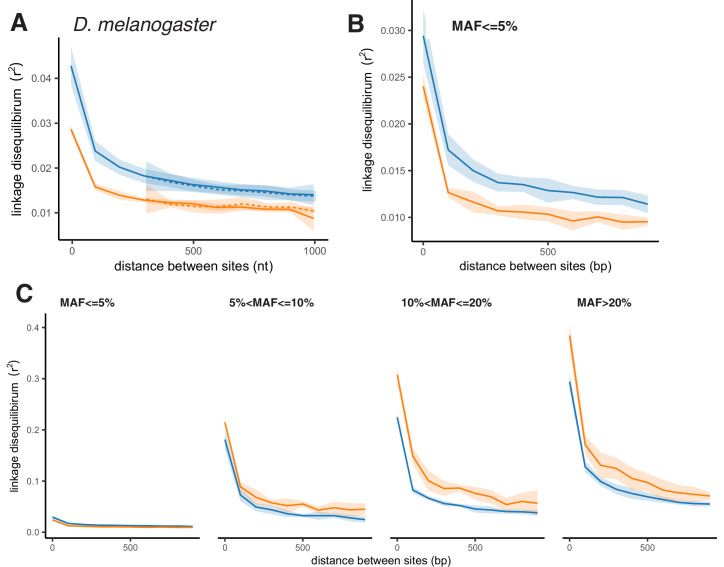
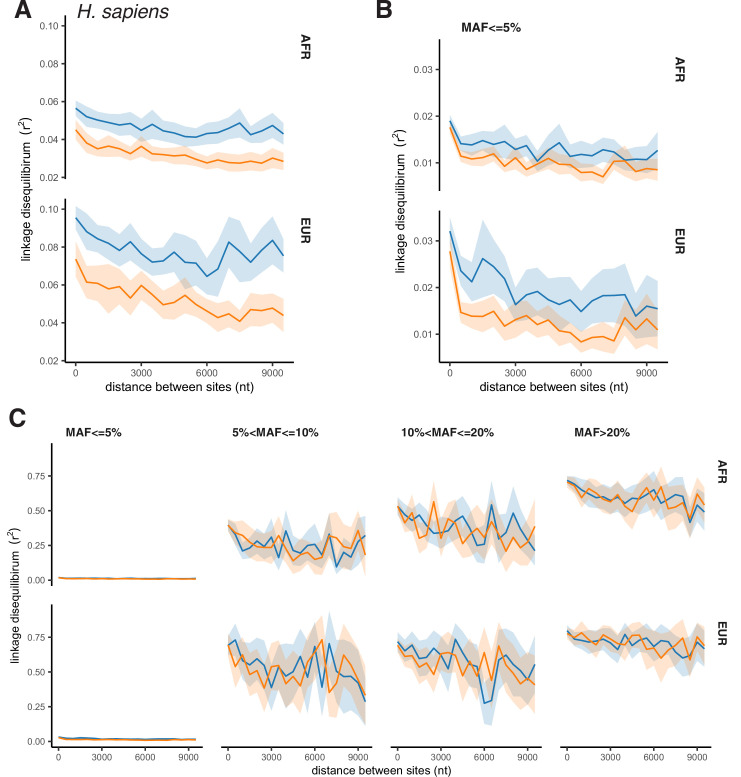
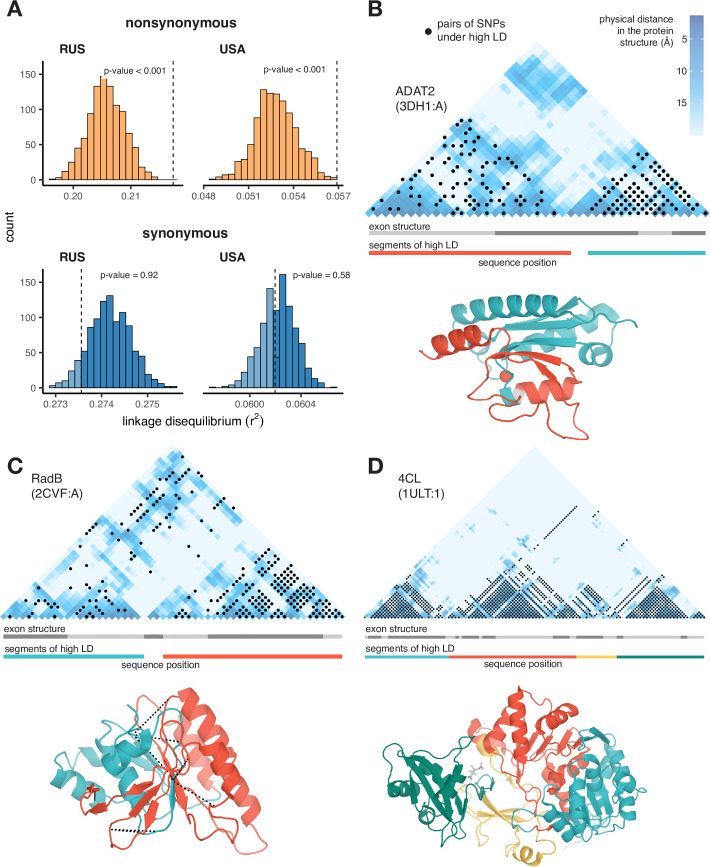
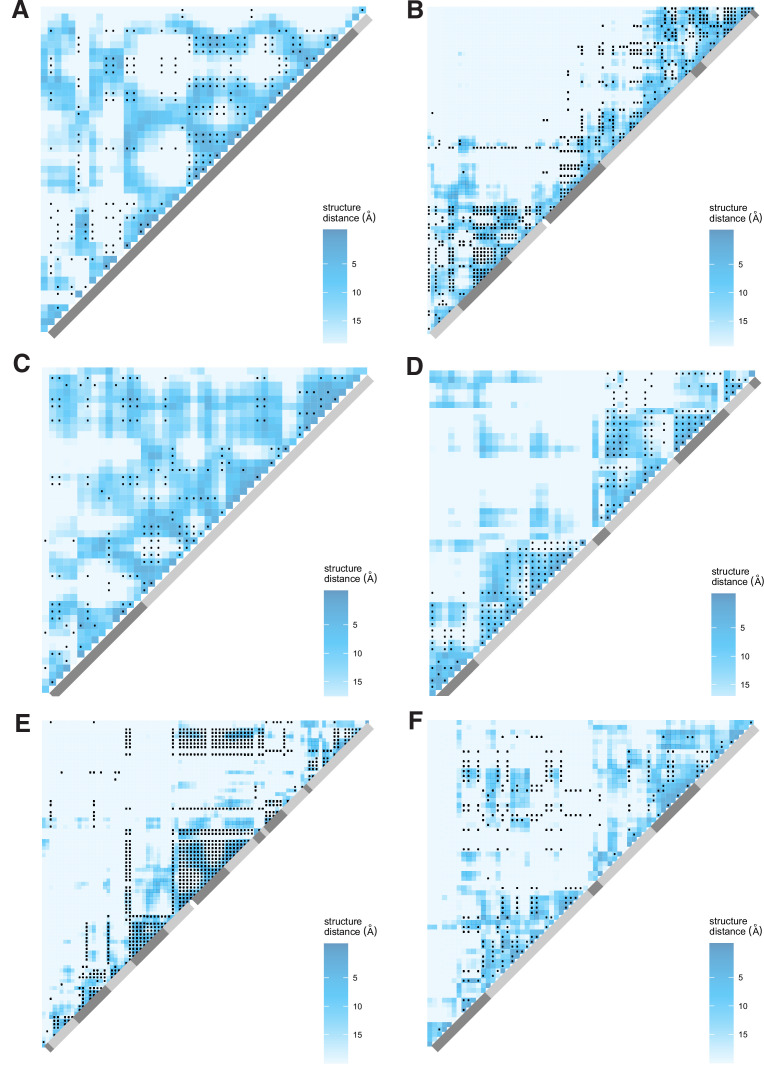
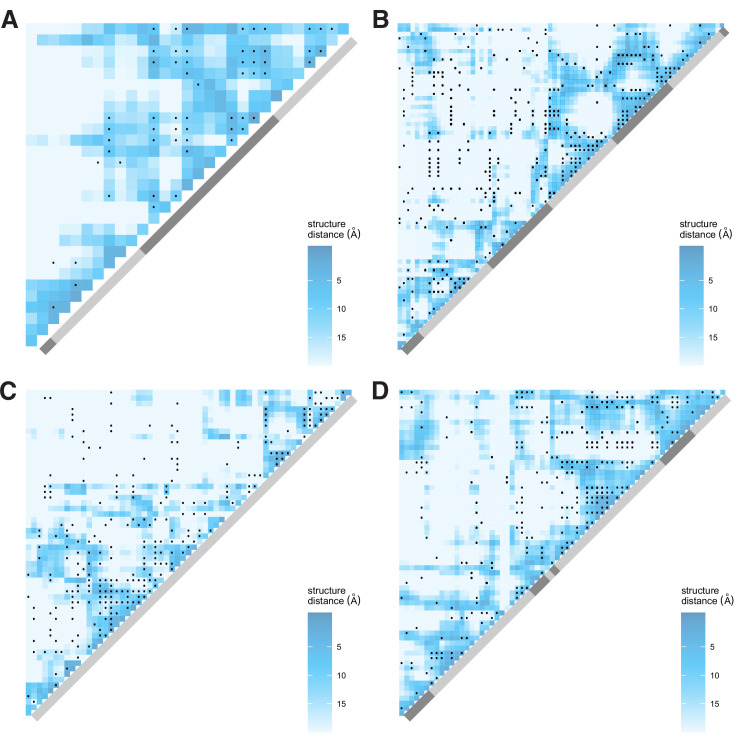
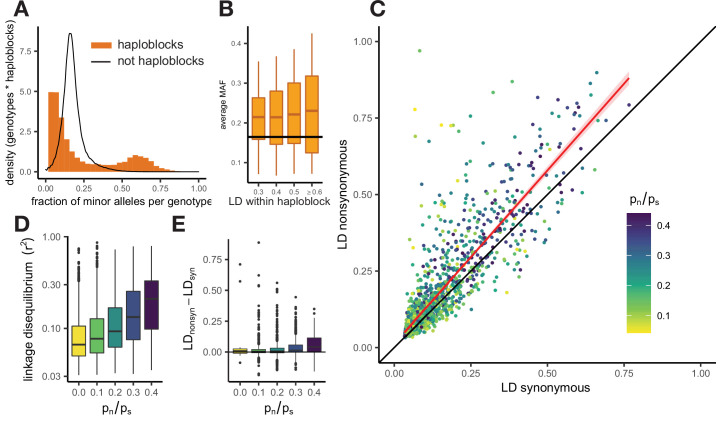
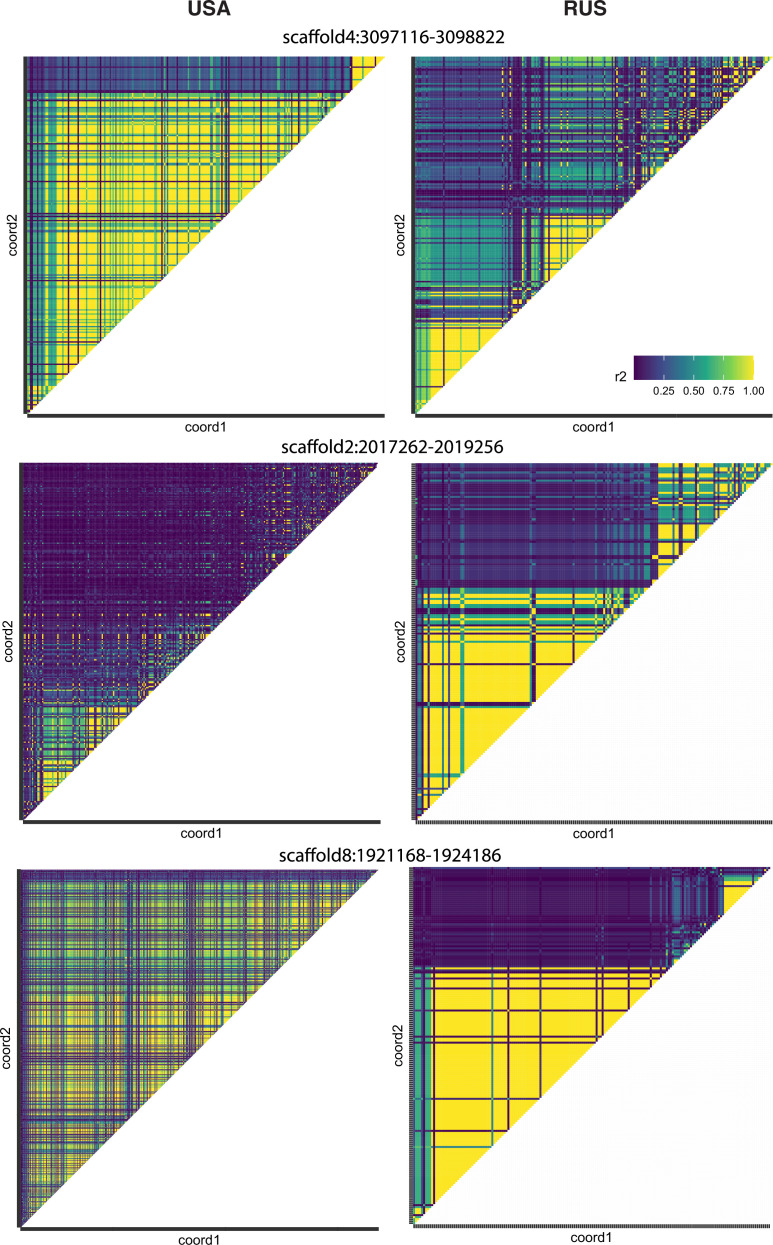
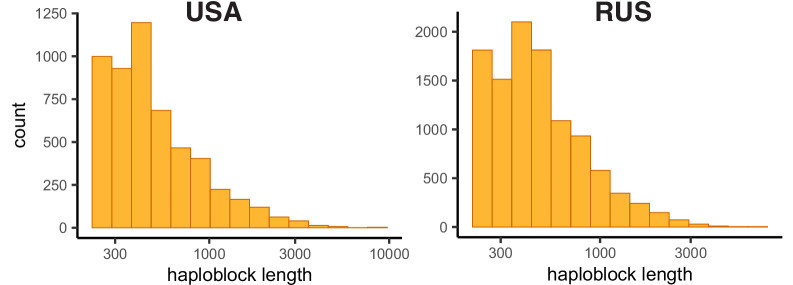
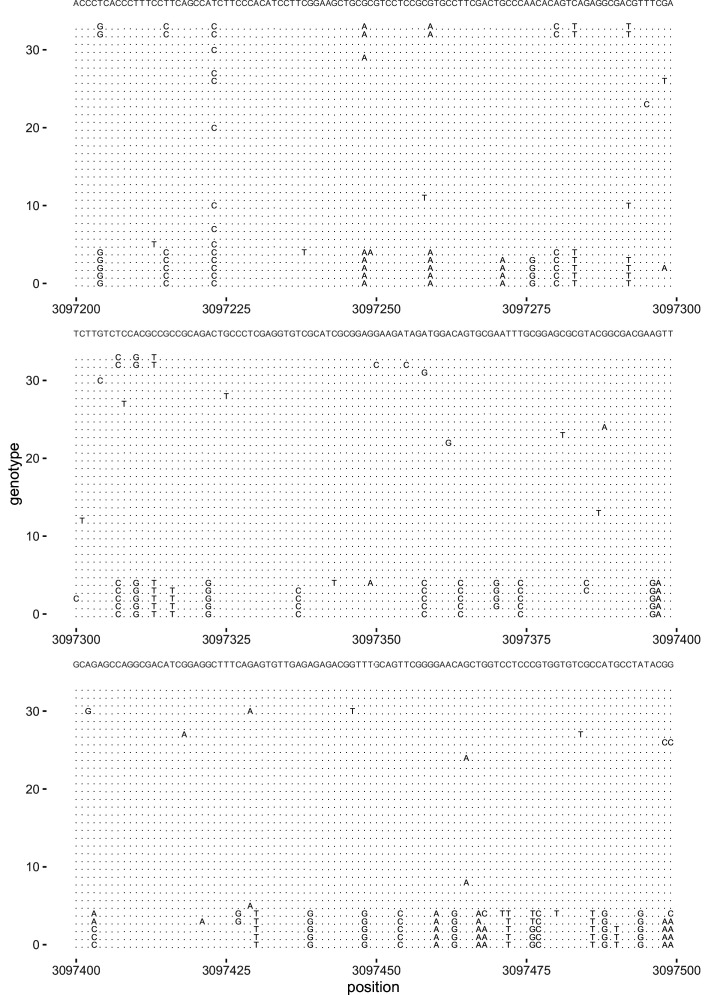
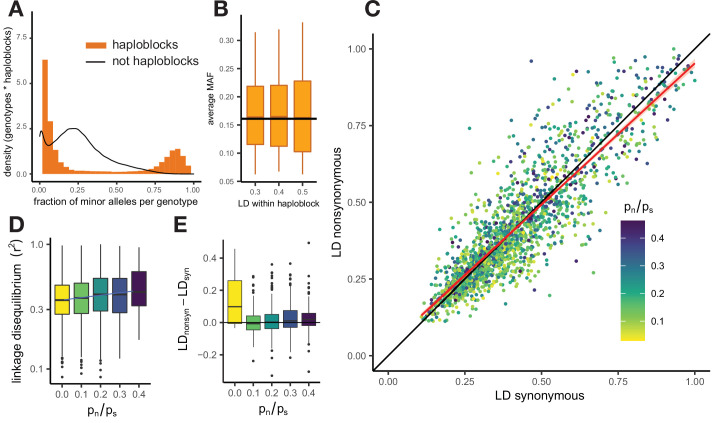
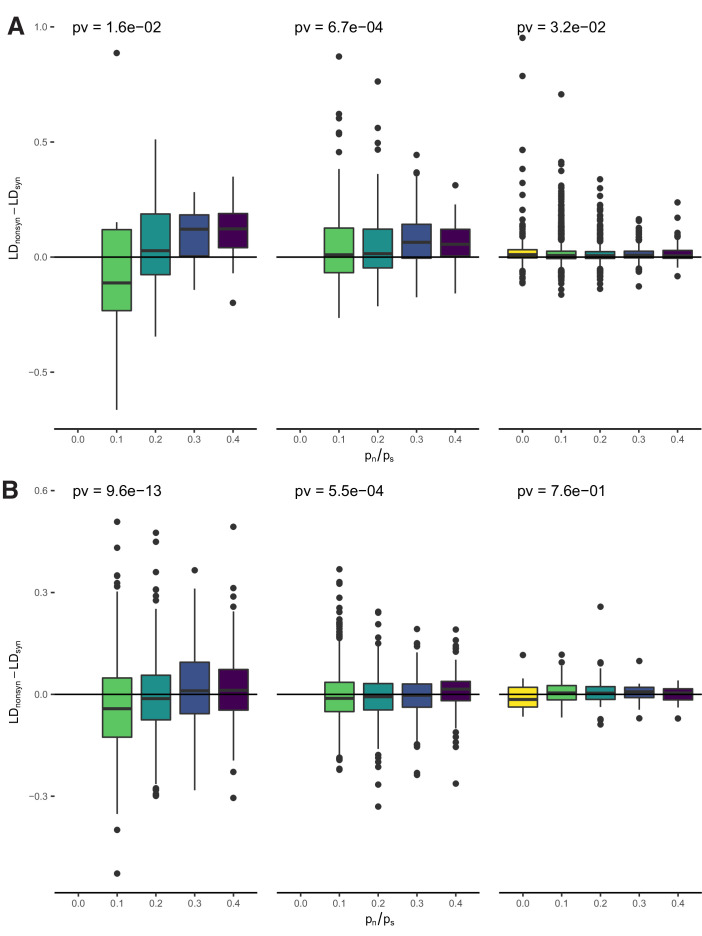
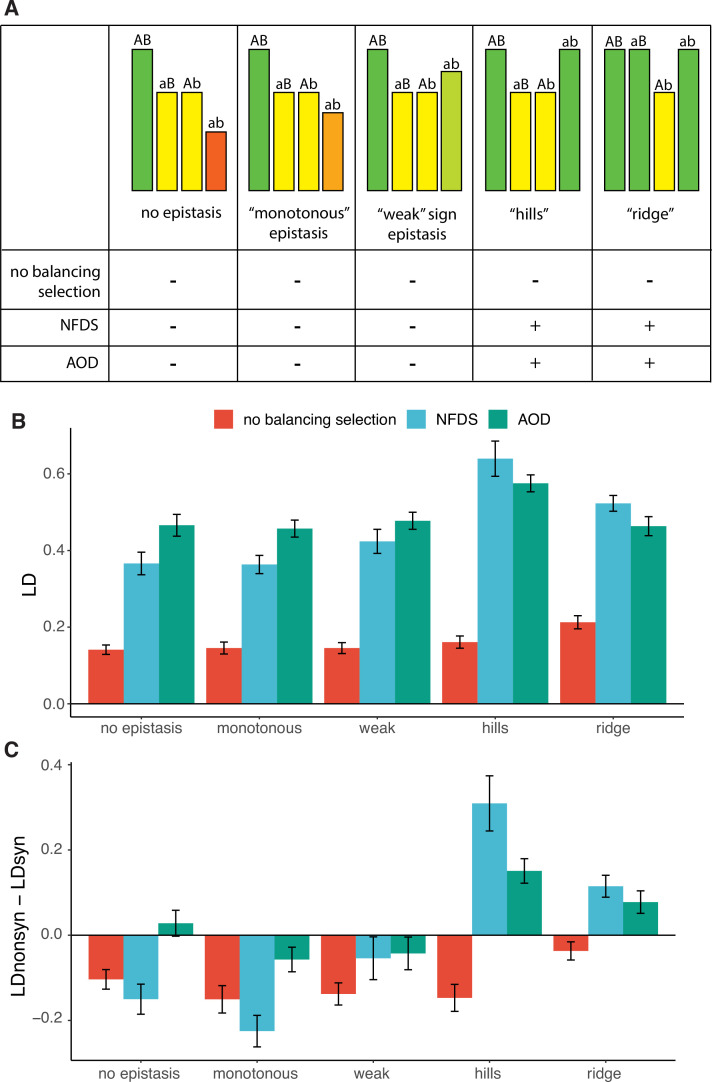
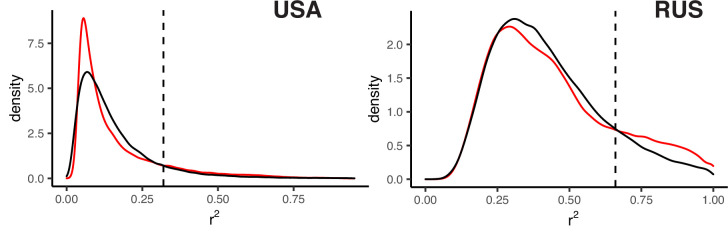
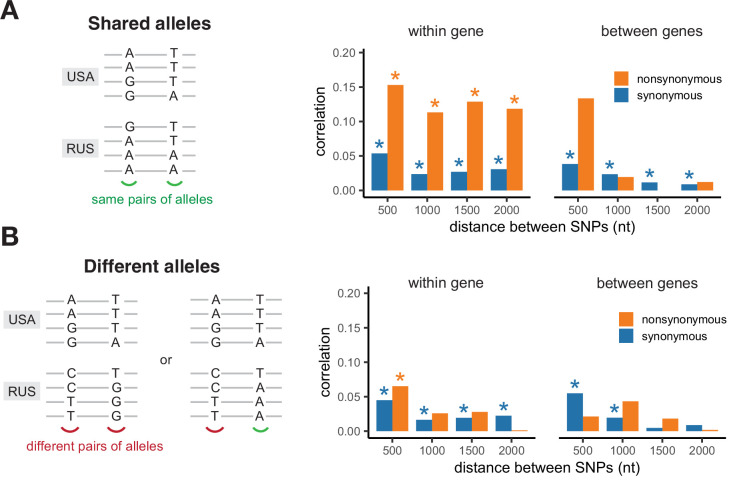
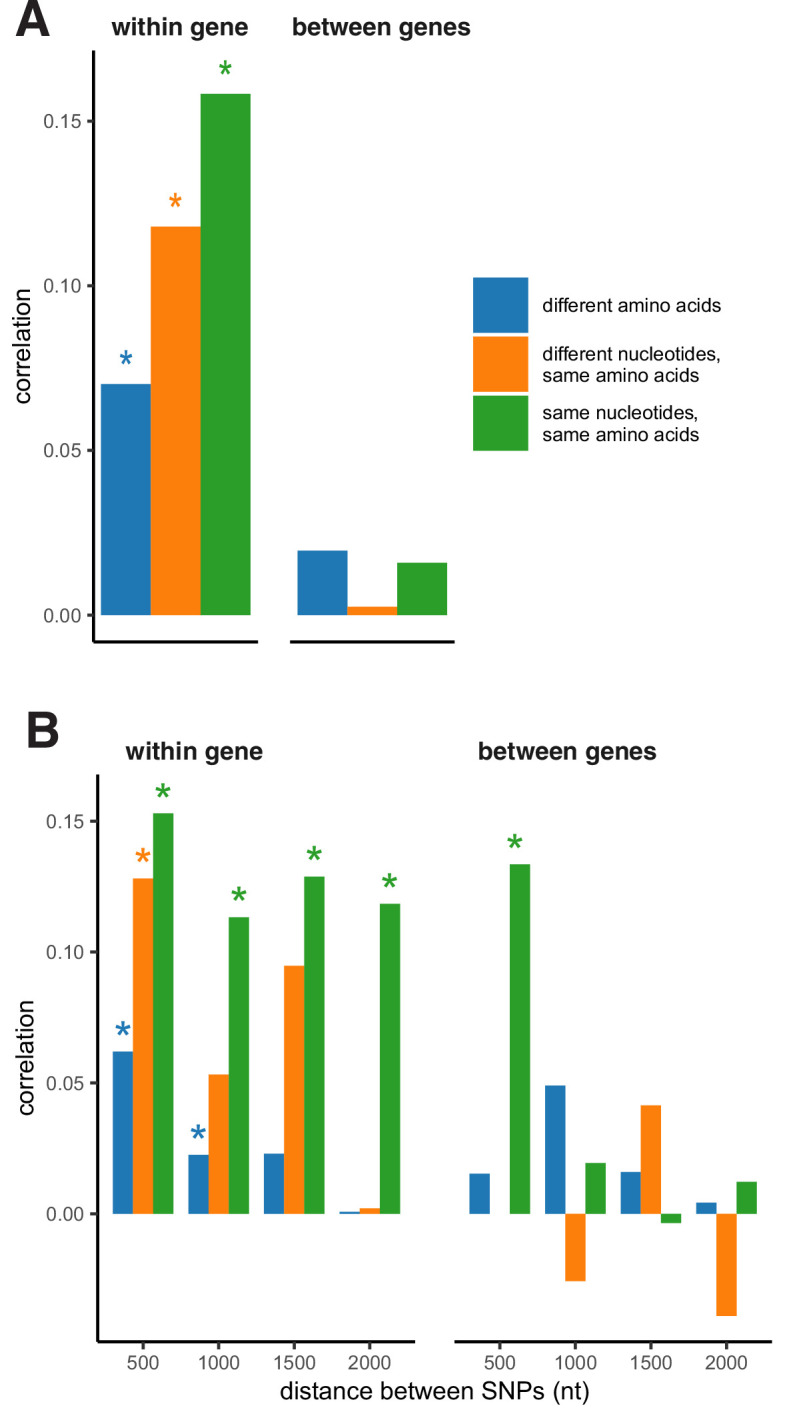
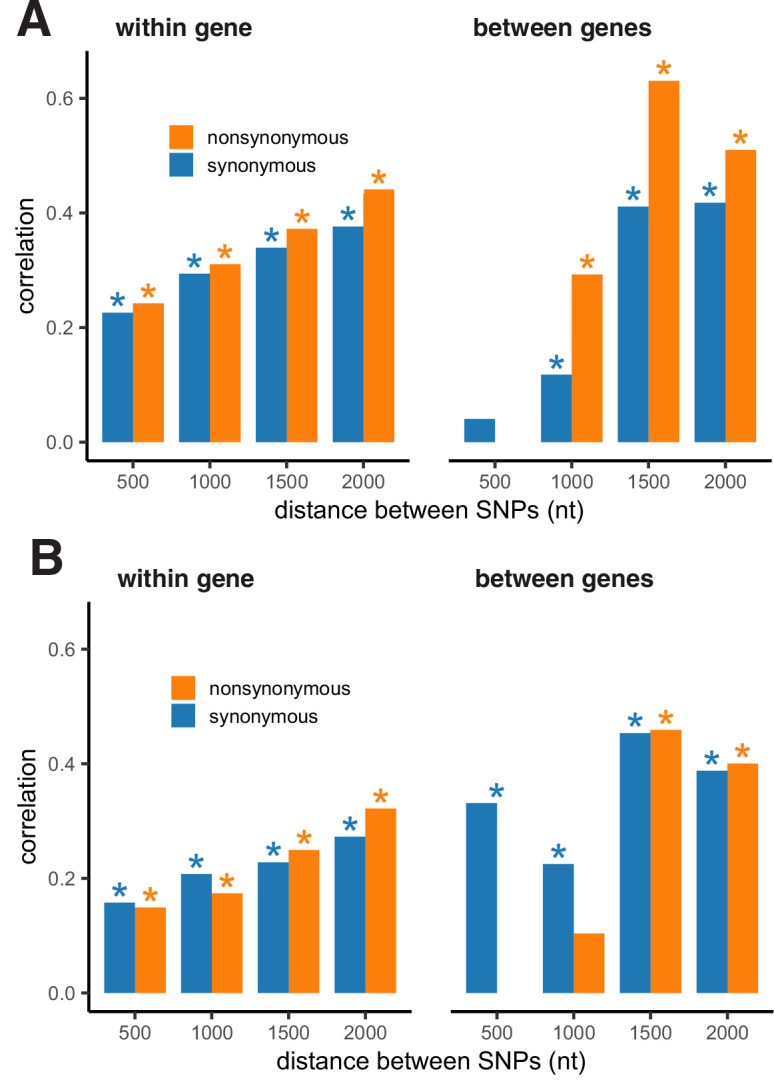
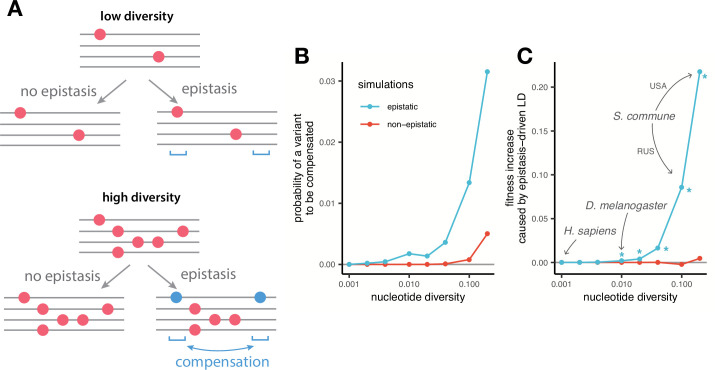
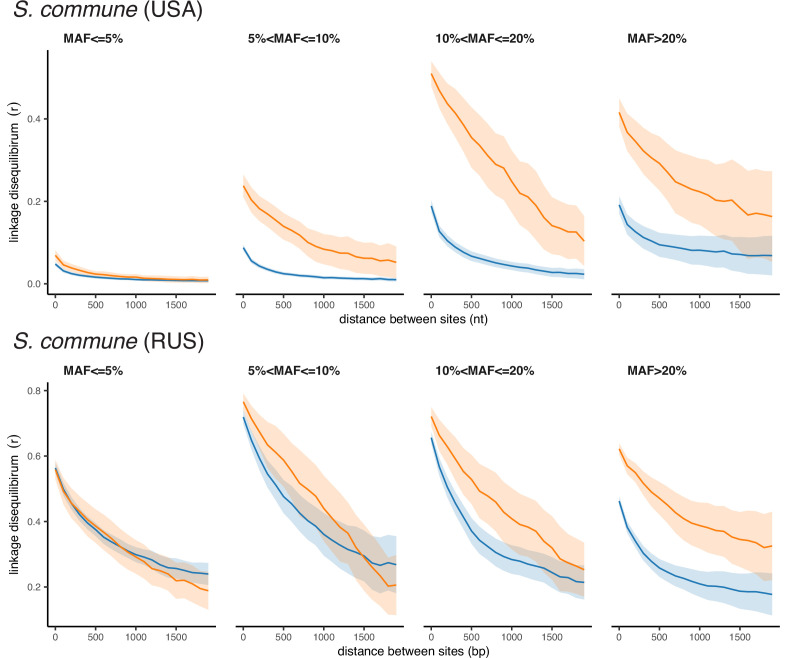
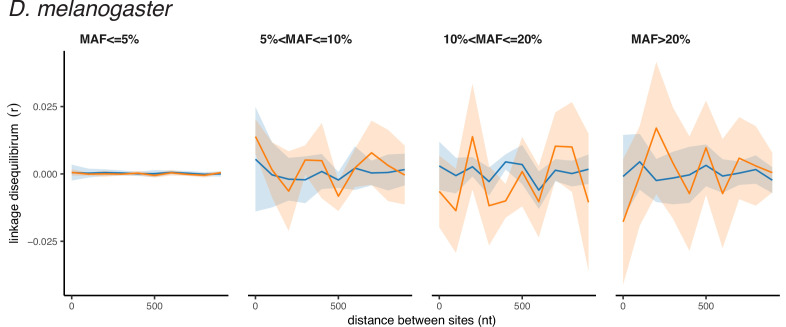
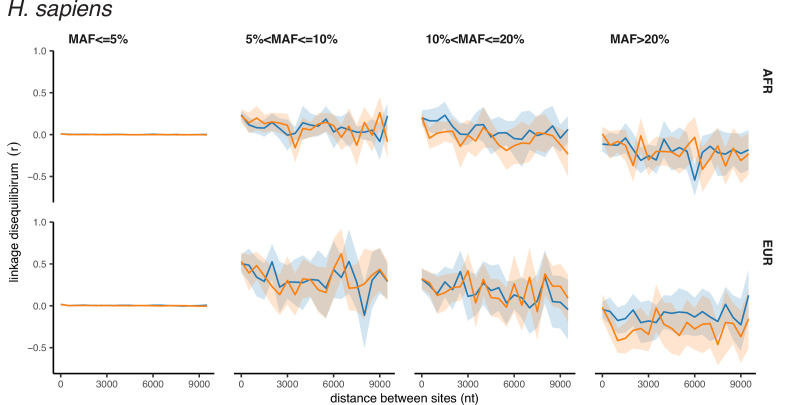
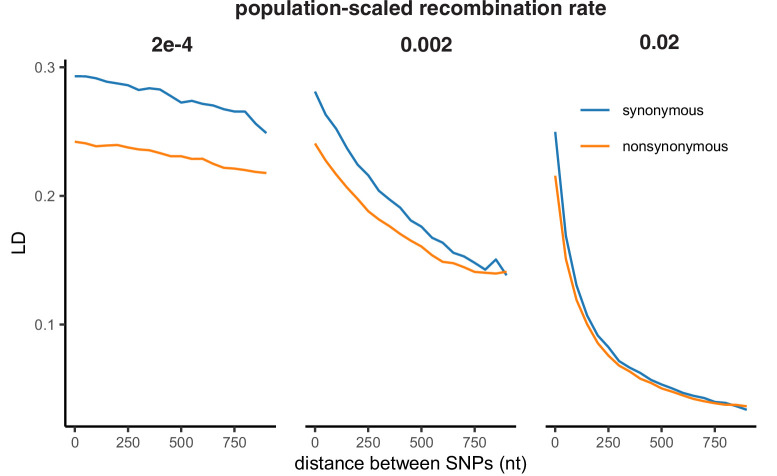
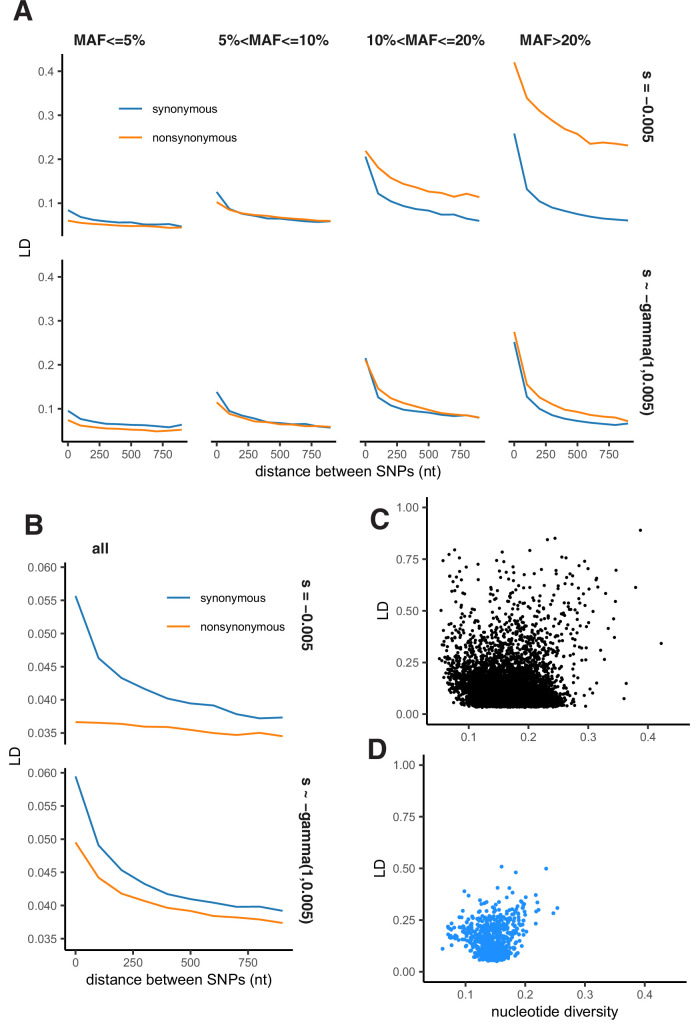
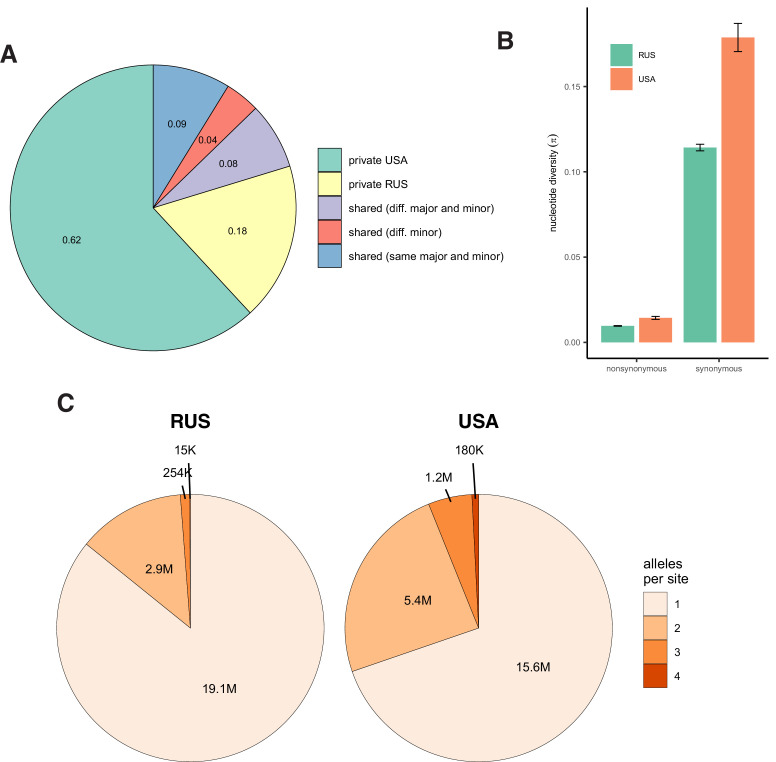
It is natural to assume that patterns of genetic variation in hyperpolymorphic species can reveal large-scale properties of the fitness landscape that are hard to detect by studying species with ordinary levels of genetic variation. Here, we study such patterns in a fungus Schizophyllum commune, the most polymorphic species known. Throughout the genome, short-range linkage disequilibrium (LD) caused by attraction of minor alleles is higher between pairs of nonsynonymous than of synonymous variants. This effect is especially pronounced for pairs of sites that are located within the same gene, especially if a large fraction of the gene is covered by haploblocks, genome segments where the gene pool consists of two highly divergent haplotypes, which is a signature of balancing selection. Haploblocks are usually shorter than 1000 nucleotides, and collectively cover about 10% of the S. commune genome. LD tends to be substantially higher for pairs of nonsynonymous variants encoding amino acids that interact within the protein. There is a substantial correlation between LDs at the same pairs of nonsynonymous mutations in the USA and the Russian populations. These patterns indicate that selection in S. commune involves positive epistasis due to compensatory interactions between nonsynonymous alleles. When less polymorphic species are studied, analogous patterns can be detected only through interspecific comparisons.
Keywords: D. melanogaster; epistasis; evolutionary biology; genetic diversity; genetics; genomics; human; linkage disequilibrium; population genetics; schizophyllum commune.
Plain language summary
Changes to DNA known as mutations may alter how the proteins and other components of a cell work, and thus play an important role in allowing living things to evolve new traits and abilities over many generations. Whether a mutation is beneficial or harmful may differ depending on the genetic background of the individual – that is, depending on other mutations present in other positions within the same gene – due to a phenomenon called epistasis. Epistasis is known to affect how various species accumulate differences in their DNA compared to each other over time. For example, a mutation that is rare in humans and known to cause disease may be widespread in other primates because its negative effect is canceled out by another mutation that is standard for these species but absent in humans. However, it remains unclear whether epistasis plays a significant part in shaping genetic differences between individuals of the same species. A type of fungus known as Schizophyllum commune lives on rotting wood and is found across the world. It is one of the most genetically diverse species currently known, so there is a higher chance of pairs of compensatory mutations occurring and persisting for a long time in S. commune than in most other species, providing a unique opportunity to study epistasis. Here, Stolyarova et al. studied two distinct populations of S. commune, one from the USA and one from Russia. The team found that – unlike in humans, flies and other less genetically diverse species – epistasis maintains combinations of mutations in S. commune that individually would be harmful to the fungus but together compensate for each other. For example, pairs of mutations affecting specific molecules known as amino acids – the building blocks of proteins – that physically interact with each other tended to be found together in the same individuals. One potential downside of having pairs of compensatory mutations in the genome is that when the organism reproduces, the process of making sex cells may split up these pairs so that harmful mutations are inherited without their partner mutations. Thus, epistasis may have helped shape the way S. commune and other genetically diverse species have evolved.
© 2022, Stolyarova et al.
Conflict of interest statement
AS, TN, EZ, AF, AK, GB No competing interests declared
Figures
References
-
- Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA. SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. Journal of Computational Biology. 2012;19:455–477. doi: 10.1089/cmb.2012.0021. - DOI - PMC - PubMed
-
- Baranova MA, Logacheva MD, Penin AA, Seplyarskiy VB, Safonova YY, Naumenko SA, Klepikova AV, Gerasimov ES, Bazykin GA, James TY, Kondrashov AS. Extraordinary Genetic Diversity in a Wood Decay Mushroom. Molecular Biology and Evolution. 2015;32:2775–2783. doi: 10.1093/molbev/msv153. - DOI - PMC - PubMed
Publication types
MeSH terms
LinkOut - more resources
Full Text Sources
Research Materials
